Molecular Characterization of Leishmania RNA virus 2 in Leishmania major from Uzbekistan



Abstract
:1. Introduction
2. Materials and Methods
2.1. Parasite Culture and RNA Isolation
2.2. dsRNA Isolation and Next-Generation Sequencing
2.3. Bioinformatics Analysis
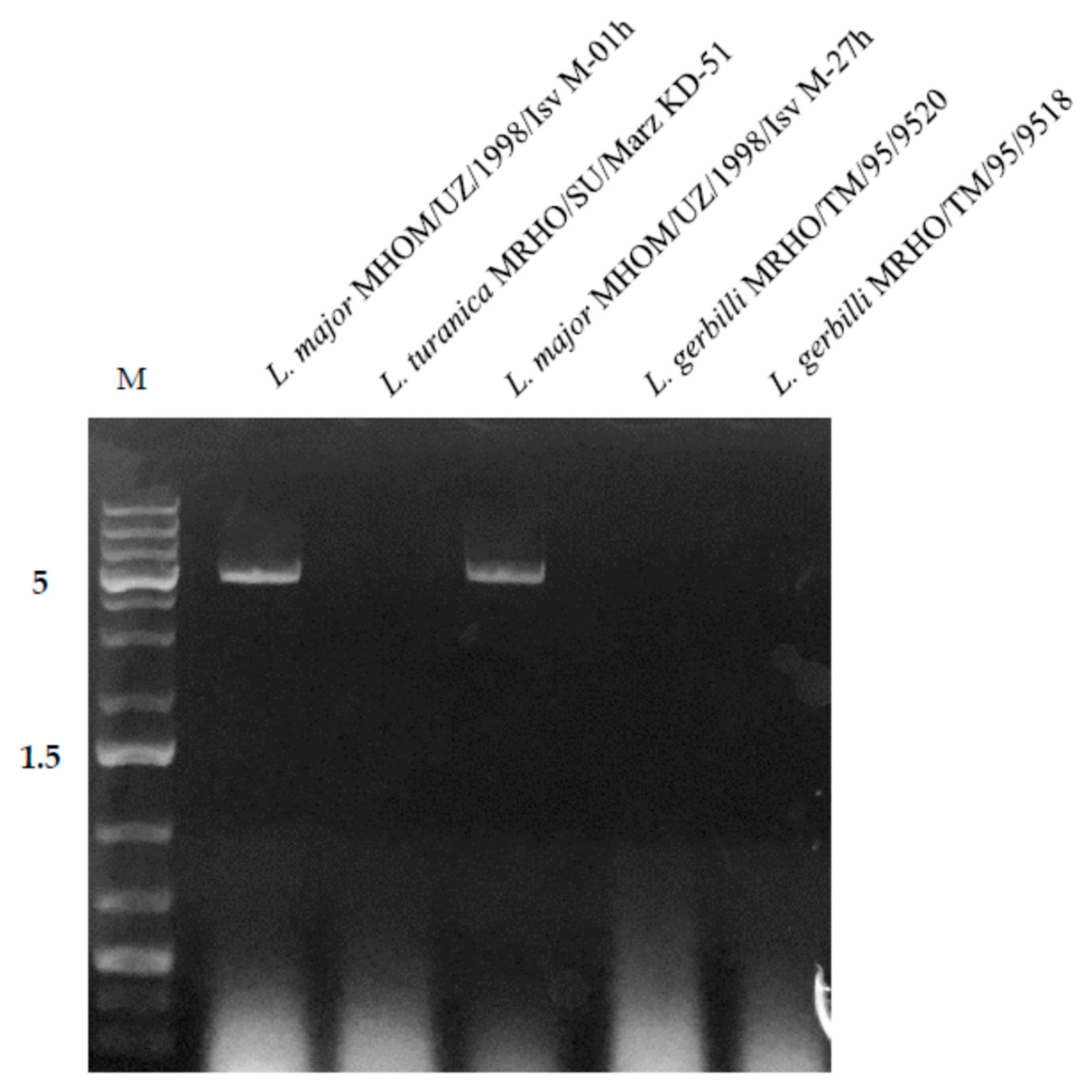
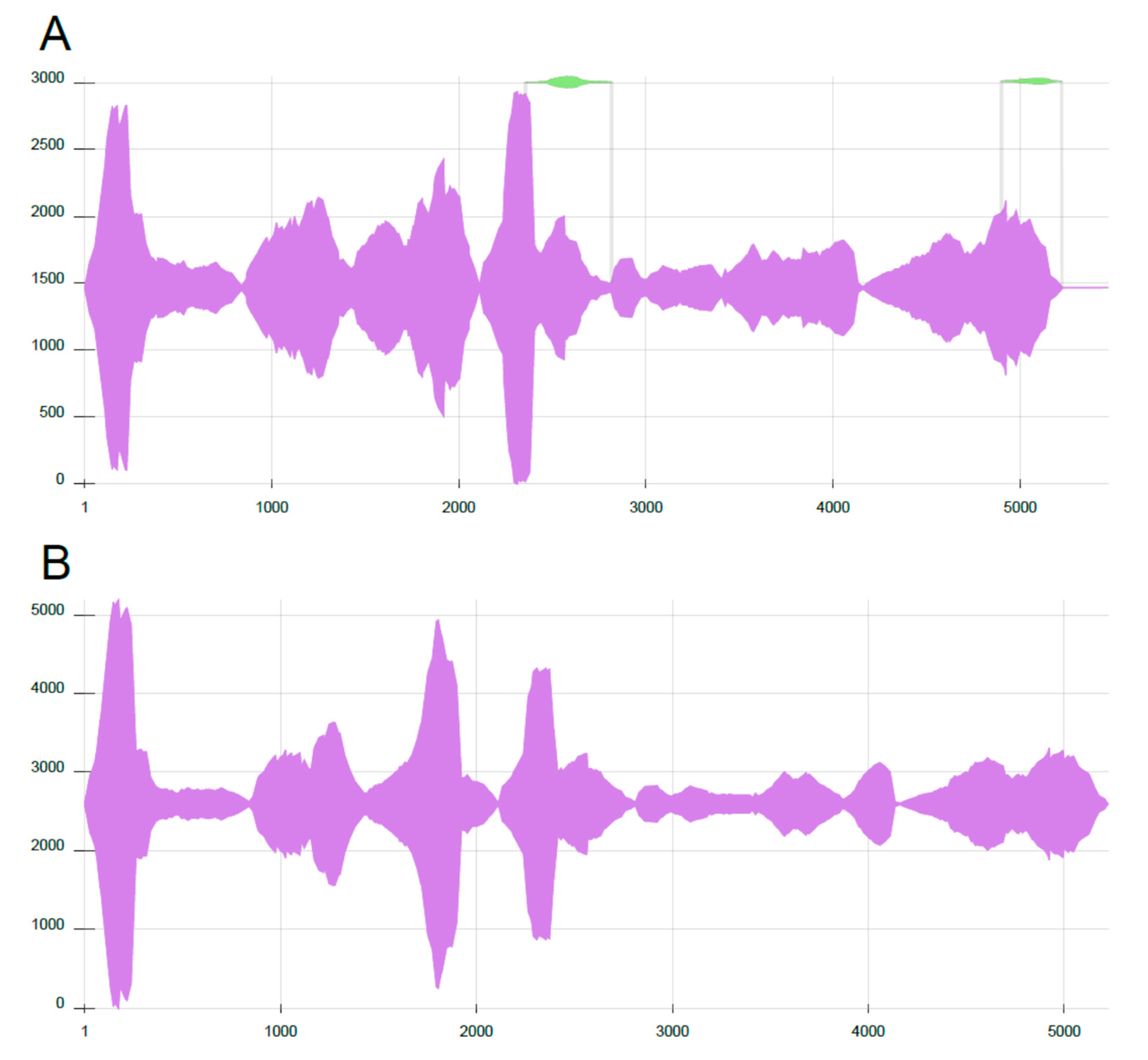
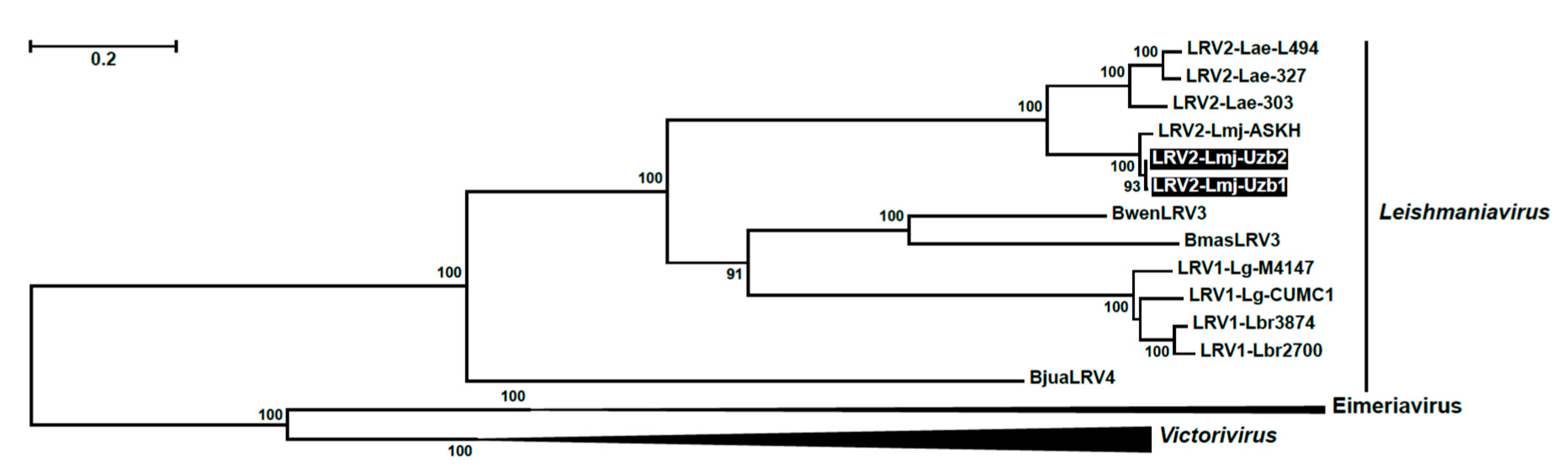
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.E.; Takagi, Y.; Parent, K.N.; Cardone, G.; Nibert, M.L.; Baker, T.S. Three-dimensional structure of a protozoal double-stranded RNA virus that infects the enteric pathogen Giardia lamblia. J. Virol. 2015, 89, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Parent, K.N.; Takagi, Y.; Cardone, G.; Olson, N.H.; Ericsson, M.; Yang, M.; Lee, Y.; Asara, J.M.; Fichorova, R.N.; Baker, T.S.; et al. Structure of a protozoan virus from the human genitourinary parasite Trichomonas vaginalis. MBio 2013. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Li, H.; Cardone, G.; Nibert, M.L.; Ghabrial, S.A.; Baker, T.S. Three-dimensional structure of victorivirus HvV190S suggests coat proteins in most totiviruses share a conserved core. PLoS Pathog. 2013, 9, e1003225. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.D.; Weeks, R.L.; Guilbride, P.J. Myler molecular organization of Leishmania RNA virus 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8596–8600. [Google Scholar] [CrossRef] [PubMed]
- Scheffter, S.; Widmer, G.; Patterson, J.L. Complete sequence of Leishmania RNA virus 1–4 and identification of conserved sequences. Virology 1994, 199, 479–483. [Google Scholar] [CrossRef]
- Lee, S.E.; Suh, J.M.; Scheffter, S.; Patterson, J.L.; Chung, I.K. Identification of a ribosomal frameshift in Leishmania RNA virus 1–4. J. Biochem. 1996, 120, 22–25. [Google Scholar] [CrossRef]
- Zangger, H.; Hailu, A.; Desponds, C.; Lye, L.F.; Akopyants, N.S.; Dobson, D.E.; Ronet, C.; Ghalib, H.; Beverley, S.M.; Fasel, N. Leishmania aethiopica field isolates bearing an endosymbiontic dsRNA virus induce pro-inflammatory cytokine response. PLoS Negl. Trop. Dis. 2014, 8, e2836. [Google Scholar] [CrossRef]
- Scheffter, S.M.; Ro, Y.T.; Chung, I.K.; Patterson, J.L. The complete sequence of Leishmania RNA virus LRV2-1, a virus of an Old World parasite strain. Virology 1995, 212, 84–90. [Google Scholar] [CrossRef]
- Widmer, G.; Dooley, S. Phylogenetic analysis of Leishmania RNA virus and Leishmania suggests ancient virus-parasite association. Nucleic Acids Res. 1995, 23, 2300–2304. [Google Scholar] [CrossRef]
- Okamoto, K.; Miyazaki, N.; Larsson, D.S.; Kobayashi, D.; Svenda, M.; Muhlig, K.; Maia, F.R.; Gunn, L.H.; Isawa, H.; Kobayashi, M.; et al. The infectious particle of insect-borne totivirus-like Omono River virus has raised ridges and lacks fibre complexes. Sci. Rep. 2016, 6, 33170. [Google Scholar] [CrossRef] [PubMed]
- Grybchuk, D.; Kostygov, A.Y.; Macedo, D.H.; Votypka, J.; Lukes, J.; Yurchenko, V. RNA viruses in Blechomonas (Trypanosomatidae) and evolution of Leishmaniavirus. MBio 2018. [Google Scholar] [CrossRef] [PubMed]
- Atayde, V.D.; da Silva, A.; Filho, L.; Chaparro, V.; Zimmermann, A.; Martel, C.; Jaramillo, M.; Olivier, M. Exploitation of the Leishmania exosomal pathway by Leishmania RNA virus 1. Nat. Microbiol. 2019, 4, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.F.; Hickerson, S.M.; et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef]
- Hartley, M.A.; Drexler, S.; Ronet, C.; Beverley, S.M.; Fasel, N. The immunological, environmental, and phylogenetic perpetrators of metastatic leishmaniasis. Trends Parasitol. 2014, 30, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Hartley, M.A.; Ronet, C.; Zangger, H.; Beverley, S.M.; Fasel, N. Leishmania RNA virus: When the host pays the toll. Front. Cell Infect Microbiol. 2012, 2, 99. [Google Scholar]
- Tirera, S.; Ginouves, M.; Donato, D.; Caballero, I.S.; Bouchier, C.; Lavergne, A.; Bourreau, E.; Mosnier, E.; Vantilcke, V.; Couppie, P.; et al. Unraveling the genetic diversity and phylogeny of Leishmania RNA virus 1 strains of infected Leishmania isolates circulating in French Guiana. PLoS Negl. Trop. Dis. 2017, 11, e0005764. [Google Scholar] [CrossRef]
- Grybchuk, D.; Kostygov, A.Y.; Macedo, D.H.; d’Avila-Levy, C.M.; Yurchenko, V. RNA viruses in trypanosomatid parasites: A historical overview. Mem. Inst. Oswaldo Cruz 2018, 113, e170487. [Google Scholar] [CrossRef]
- Grybchuk, D.; Akopyants, N.S.; Kostygov, A.Y.; Konovalovas, A.; Lye, L.F.; Dobson, D.E.; Zangger, H.; Fasel, N.; Butenko, A.; Frolov, A.O.; et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. USA. 2018, 115, E506–E515. [Google Scholar] [CrossRef]
- Adaui, V.; Lye, L.F.; Akopyants, N.S.; Zimic, M.; Llanos-Cuentas, A.; Garcia, L.; Maes, I.; De Doncker, S.; Dobson, D.E.; Arevalo, J.; et al. Association of the endobiont double-stranded RNA virus LRV1 with treatment failure for human leishmaniasis caused by Leishmania braziliensis in Peru and Bolivia. J. Infect. Dis. 2016, 213, 112–121. [Google Scholar] [CrossRef]
- Ginouvès, M.; Simon, S.; Bourreau, E.; Lacoste, V.; Ronet, C.; Couppie, P.; Nacher, M.; Demar, M.; Prevot, G. Prevalence and distribution of Leishmania RNA Virus 1 in Leishmania parasites from French Guiana. Am. J. Trop. Med. Hyg. 2016, 94, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Castiglioni, P.; Hartley, M.A.; Eren, R.O.; Prevel, F.; Desponds, C.; Utzschneider, D.T.; Zehn, D.; Cusi, M.G.; Kuhlmann, F.M.; et al. Type I interferons induced by endogenous or exogenous viral infections promote metastasis and relapse of leishmaniasis. Proc. Natl. Acad. Sci. USA. 2017, 114, 4987–4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paranaiba, L.F.; Pinheiro, L.J.; Macedo, D.H.; Menezes-Neto, A.; Torrecilhas, A.C.; Tafuri, W.L.; Soares, R.P. An overview on Leishmania (Mundinia) enriettii: Biology, immunopathology, LRV and extracellular vesicles during the host-parasite interaction. Parasitology 2018, 145, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.; Gradoni, L. The Leishmaniases: Old Neglected Tropical Diseases; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Sukla, S.; Roy, S.; Sundar, S.; Biswas, S. Leptomonas seymouri narna-like virus 1 and not leishmaniaviruses detected in kala-azar samples from India. Arch. Virol. 2017, 162, 3827–3835. [Google Scholar] [CrossRef]
- Kraeva, N.; Butenko, A.; Hlaváčová, J.; Kostygov, A.; Myškova, J.; Grybchuk, D.; Leštinová, T.; Votýpka, J.; Volf, P.; Opperdoes, F.; et al. Leptomonas seymouri: Adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with Leishmania donovani. PLoS Pathog. 2015, 11, e1005127. [Google Scholar] [CrossRef]
- Lye, L.F.; Akopyants, N.S.; Dobson, D.E.; Beverley, S.M. A Narnavirus-like element from the trypanosomatid protozoan parasite Leptomonas seymouri. Genome Announc. 2016. [Google Scholar] [CrossRef]
- Hajjaran, H.; Mahdi, M.; Mohebali, M.; Samimi-Rad, K.; Ataei-Pirkooh, A.; Kazemi-Rad, E.; Naddaf, S.R.; Raoofian, R. Detection and molecular identification of Leishmania RNA virus (LRV) in Iranian Leishmania species. Arch. Virol. 2016, 161, 3385–3390. [Google Scholar] [CrossRef]
- Kurt, O.; Mansur, N.; Cavus, I.; Ozcan, O.; Batir, M.B.; Gunduz, C.; Sezerman, O.U.; Ozbilgin, A. First report and in silico analysis of Leishmania virus (LRV2) identified in an autochthonous Leishmania major isolate in Turkey. New Microbiol. 2019, 42, 64–67. [Google Scholar]
- Faulde, M.K.; Werner, A.; Heyl, G. Untreated zoonotic cutaneous leishmaniasis characterizing a highly aggressive strain type of Leishmania major in Uzbekistan. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1432–1433. [Google Scholar] [CrossRef]
- Zhirenkina, E.N.; Ponirovskii, E.N.; Strelkova, M.V.; Morozov, E.N.; Flegontov, P.N.; Kolesnikov, A.A.; Ponomareva, V.I.; Nasyrova, R.M.; Kovalenko, D.A.; Fatullaeva, A.A.; et al. The epidemiological features of visceral leishmaniasis, revealed on examination of children by Polymerase Chain Reaction, in the Papsky District, Namangan Region, Uzbekistan. Med. Parazitol. Parazit. Bolezn. 2011, 37–41. (in Russian). [Google Scholar]
- Strelkova, M.V.; Ponirovsky, E.N.; Morozov, E.N.; Zhirenkina, E.N.; Razakov, S.A.; Kovalenko, D.A.; Schnur, L.F.; Schonian, G. A narrative review of visceral leishmaniasis in Armenia, Azerbaijan, Georgia, Kazakhstan, Kyrgyzstan, Tajikistan, Turkmenistan, Uzbekistan, the Crimean Peninsula and Southern Russia. Parasit. Vectors 2015, 8, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chajbullinova, A.; Votýpka, J.; Sádlová, J.; Kvapilová, K.; Seblová, V.; Kreisinger, J.; Jirků, M.; Sanjoba, C.; Gantuya, S.; Matsumoto, Y.; et al. The development of Leishmania turanica in sand flies and competition with L. major. Parasit. Vectors 2012, 5, 219. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.S. Genome Project Data Processing The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008, 36, D13–D21. [Google Scholar] [CrossRef]
- Karagiannis, K.; Simonyan, V.; Chumakov, K.; Mazumder, R. Separation and assembly of deep sequencing data into discrete sub-population genomes. Nucleic Acids Res. 2017, 45, 10989–11003. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votýpka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trends Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef]
- Zangger, H.; Ronet, C.; Desponds, C.; Kuhlmann, F.M.; Robinson, J.; Hartley, M.A.; Prevel, F.; Castiglioni, P.; Pratlong, F.; Bastien, P.; et al. Detection of Leishmania RNA virus in Leishmania parasites. PLoS Negl. Trop. Dis. 2013, 7, e2006. [Google Scholar] [CrossRef]
- Lye, L.F.; Owens, K.; Shi, H.; Murta, S.M.; Vieira, A.C.; Turco, S.J.; Tschudi, C.; Ullu, E.; Beverley, S.M. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010, 6, e1001161. [Google Scholar] [CrossRef]
- Matveyev, A.V.; Alves, J.M.; Serrano, M.G.; Lee, V.; Lara, A.M.; Barton, W.A.; Costa-Martins, A.G.; Beverley, S.M.; Camargo, E.P.; Teixeira, M.M.; et al. The evolutionary loss of RNAi key determinants in kinetoplastids as a multiple sporadic phenomenon. J. Mol. Evol. 2017, 84, 104–115. [Google Scholar] [CrossRef]
- Brettmann, E.A.; Shaik, J.S.; Zangger, H.; Lye, L.F.; Kuhlmann, F.M.; Akopyants, N.S.; Oschwald, D.M.; Owens, K.L.; Hickerson, S.M.; Ronet, C.; et al. Tilting the balance between RNA interference and replication eradicates Leishmania RNA virus 1 and mitigates the inflammatory response. Proc. Natl. Acad. Sci. USA 2016, 113, 11998–12005. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Leishmania spp. | WHO Code | LRV2 | Source 1 | Origin |
|---|---|---|---|---|
| L. turanica | MRHO/KZ/87/MARZBK7 | - | R. opimus | Embi, Aktobe region, Kazakhstan |
| L. major | MHOM/UZ/1998/Isv M-01h | + | H. sapiens | Muborak, Qashqadaryo region, Uzbekistan |
| L. turanica | MRHO/UZ/87MarzKK-52R | - | R. opimus | Karakalpak, Jambyl region, Uzbekistan |
| L. turanica | MRHO/SU/Marz KD-51 | - | R. opimus | Karakalpak, Jambyl region, Uzbekistan |
| L. gerbilli | MRHO/TM/95/9520 | - | R. opimus | Serdar, Balkan region, Turkmenistan |
| L. gerbilli | MRHO/TM/95/9518 | - | R. opimus | Serdar, Balkan region, Turkmenistan |
| L. major | MHOM/UZ/1998/Isv M-27h | + | H. sapiens | Muborak, Qashqadaryo region, Uzbekistan |
| L. infantum | MHOM/KZ/75/MarzDzha | - | H. sapiens | Karakalpak, Jambyl region, Uzbekistan |
| L. turanica | I/TM/95/Ph-82 | - | P. papatasi | Tejen, Ahal region, Turkmenistan |
| L. major | MRHO/UZ/2003/Isv T-38g | - | R. opimus | Termez, Surxondaryo region, Uzbekistan |
| Position, nt | LRV2-Lmj-Uzb1 | LRV2-Lmj-Uzb1 | Frequency, % | Coverage |
|---|---|---|---|---|
| 389 | C | T | 100 | 384 |
| 539 | T | A | 40 | 372 |
| 1181 | C | A | 100 | 1525 |
| 1415 | T | C | 52 | 430 |
| 1517 | C | T | 100 | 732 |
| 1772 | A | G | 100 | 3616 |
| 1835 | A | G | 100 | 4188 |
| 1892 | G | A | 100 | 3890 |
| 1922 | T | G | 100 | 1434 |
| 2603 | T | A | 100 | 836 |
| 2648 | G | A | 100 | 593 |
| 3064 | T | C | 100 | 345 |
| 3125 | G | A | 100 | 493 |
| 3434 | A | G | 31 | 224 |
| 3464 | A | G | 100 | 289 |
| 3806 | A | T | 100 | 698 |
| 3809 | G | A | 100 | 686 |
| 3881 | G | A | 100 | 704 |
| 4892 | C | T | 100 | 1154 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleschenko, Y.; Grybchuk, D.; Matveeva, N.S.; Macedo, D.H.; Ponirovsky, E.N.; Lukashev, A.N.; Yurchenko, V. Molecular Characterization of Leishmania RNA virus 2 in Leishmania major from Uzbekistan. Genes 2019, 10, 830. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100830
Kleschenko Y, Grybchuk D, Matveeva NS, Macedo DH, Ponirovsky EN, Lukashev AN, Yurchenko V. Molecular Characterization of Leishmania RNA virus 2 in Leishmania major from Uzbekistan. Genes. 2019; 10(10):830. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100830
Chicago/Turabian StyleKleschenko, Yuliya, Danyil Grybchuk, Nadezhda S. Matveeva, Diego H. Macedo, Evgeny N. Ponirovsky, Alexander N. Lukashev, and Vyacheslav Yurchenko. 2019. "Molecular Characterization of Leishmania RNA virus 2 in Leishmania major from Uzbekistan" Genes 10, no. 10: 830. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100830