GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Participant Selection
2.3. Molecular Analysis
2.4. Data Analysis
3. Results
3.1. Participant Demographics
3.2. Audiometric Characteristics
3.3. Inheritance Pattern
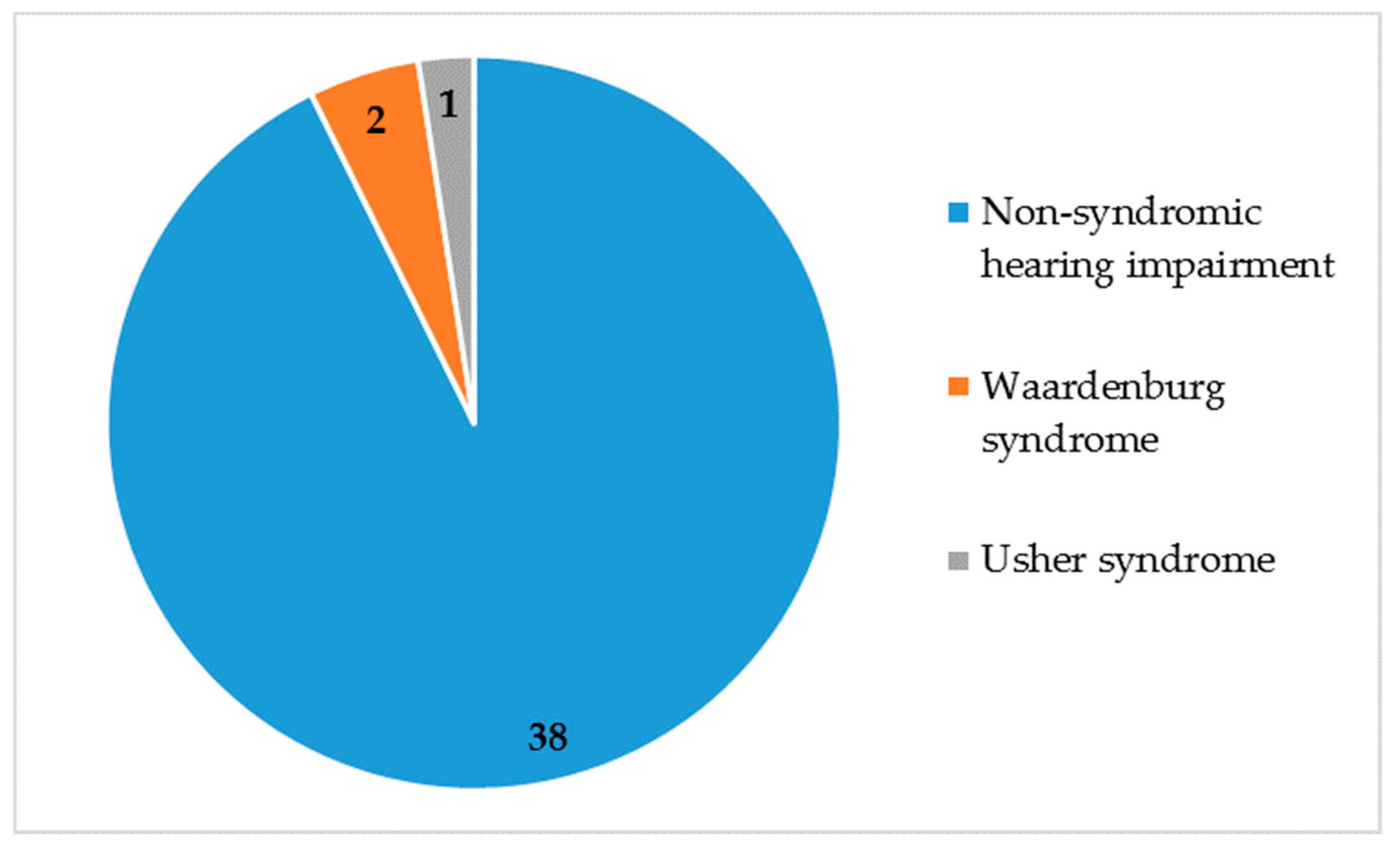
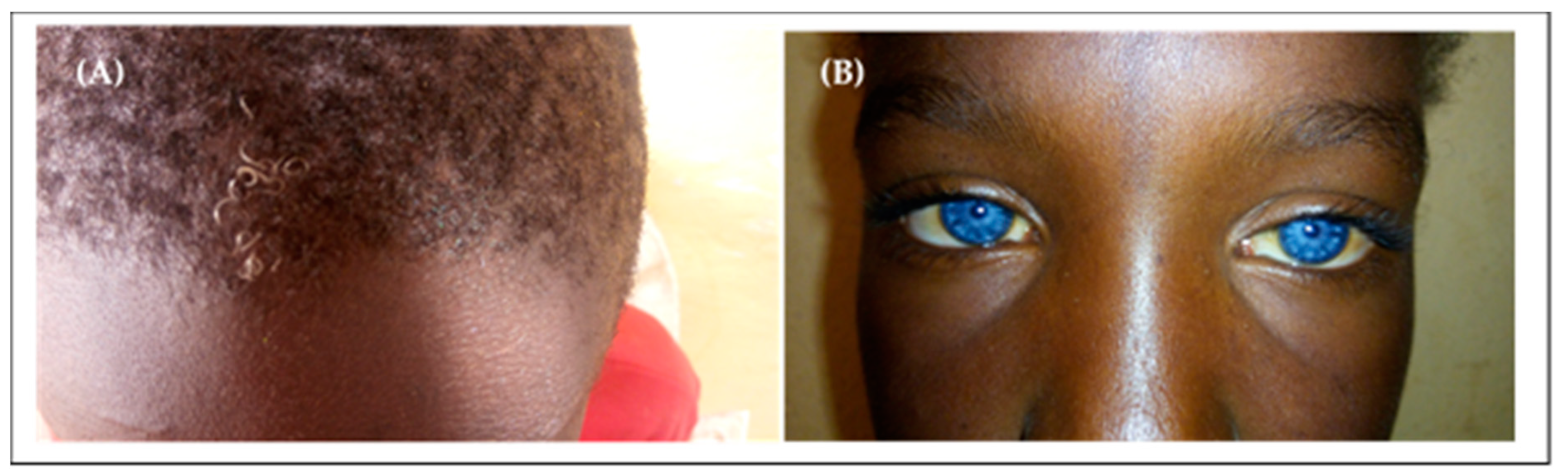
3.4. Non-Syndromic and Syndromic Hearing Impairment
3.5. Molecular Analysis Results of GJB2 and GJB6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- James, M.; Praveen, K.P.; Ninan, P.J. A study on prevalence and risk factors of hearing impairment among newborns. Int. J. Contemp. Pediatrics 2018, 5, 304–309. [Google Scholar] [CrossRef]
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lebeko, K.; Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Wonkam, A. Genetics of hearing loss in Africans: Use of next generation sequencing is the best way forward. Pan Afr. Med. J. 2015, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. Rev. Mutat. Res. 2009, 681, 189–196. [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wang, S.; Chen, S.; Wen, Y.; Liu, B.; Xie, W.; Li, D.; Liu, L.; Huang, X.; Sun, Y.; et al. Autosomal Recessive Congenital Sensorineural Hearing Loss due to a Novel Compound Heterozygous PTPRQ Mutation in a Chinese Family. Neural Plast. 2018, 2018, 9425725. [Google Scholar] [CrossRef]
- Adhikary, B.; Ghosh, S.; Paul, S.; Bankura, B.; Pattanayak, A.K.; Biswas, S.; Maity, B.; Das, M. Spectrum and frequency of GJB2, GJB6 and SLC26A4 gene mutations among nonsyndromic hearing loss patients in eastern part of India. Gene 2015, 573, 239–245. [Google Scholar] [CrossRef]
- Welcome to the Hereditary Hearing Loss Homepage|Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 19 November 2018).
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- del Castillo, I.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and Evolutionary Origins of the del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing-Impaired Subjects: A Multicenter Study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Population, Total|Data. Available online: https://data.worldbank.org/indicator/SP.POP.TOTL (accessed on 5 April 2019).
- Wonkam, A.; Noubiap, J.J.N.; Djomou, F.; Fieggen, K.; Njock, R.; Toure, G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med Genet. 2013, 56, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Friedlaender, F.R.; Ehret, C.; Ranciaro, A.; Froment, A.; Hirbo, J.B.; Awomoyi, A.A.; Bodo, J.-M.; Doumbo, O.; et al. The Genetic Structure and History of Africans and African Americans. Science 2009, 324, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, L.; Iacona, E.; Primignani, P.; Castorina, P.; Radaelli, C.; Bo, L.D.; Coviello, D.; Ambrosetti, U. GJB2 and MTRNR1 contributions in children with hearing impairment from Northern Cameroon. Int. J. Audiol. 2011, 50, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Makubalo, N.; Wright, G.; Entfellner, J.-B.D.; Tiffin, N.; Wonkam, A. Sequencing of GJB2 in Cameroonians and Black South Africans and comparison to 1000 Genomes Project Data Support Need to Revise Strategy for Discovery of Nonsyndromic Deafness Genes in Africans. OMICS 2014, 18, 705–710. [Google Scholar] [CrossRef]
- Bosch, J.; Lebeko, K.; Nziale, J.J.N.; Dandara, C.; Makubalo, N.; Wonkam, A. In Search of Genetic Markers for Nonsyndromic Deafness in Africa: A Study in Cameroonians and Black South Africans with the GJB6 and GJA1 Candidate Genes. OMICS 2014, 18, 481–485. [Google Scholar] [CrossRef]
- BIAP—Bureau International d’Audiophonologie. Available online: https://www.biap.org/en/component/content/article/65-recommendations/ct-2-classification/5-biap-recommendation-021-bis (accessed on 18 November 2018).
- Chen, P.; Chen, H.; Fu, S.; Chen, G.; Dong, J. Prevalence of GJB6 mutations in Chinese patients with non-syndromic hearing loss. Int. J. Pediatric Otorhinolaryngol. 2012, 76, 265–267. [Google Scholar] [CrossRef]
- Pampanos, A.; Economides, J.; Iliadou, V.; Neou, P.; Leotsakos, P.; Voyiatzis, N.; Eleftheriades, N.; Tsakanikos, M.; Antoniadi, T.; Hatzaki, A.; et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int. J. Pediatr. Otorhinolaryngol. 2002, 65, 101–108. [Google Scholar] [CrossRef]
- Dahl, H.H.; Saunders, K.; Kelly, T.M.; Osborn, A.H.; Wilcox, S.; Cone-Wesson, B.; Wunderlich, J.L.; Du Sart, D.; Kamarinos, M.; Gardner, R.J.; et al. Prevalence and nature of connexin 26 mutations in children with non-syndromic deafness. Med. J. Aust. 2001, 175, 191–194. [Google Scholar] [CrossRef]
- Neocleous, V.; Aspris, A.; Shahpenterian, V.; Nicolaou, V.; Panagi, C.; Ioannou, I.; Kyamides, Y.; Anastasiadou, V.; Phylactou, L.A. High frequency of 35delG GJB2 mutation and absence of del(GJB6-D13S1830) in Greek Cypriot patients with nonsyndromic hearing loss. Genet. Test. 2006, 10, 285–289. [Google Scholar] [CrossRef]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.-Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatric Otorhinolaryngol. 2011, 75, 611. [Google Scholar] [CrossRef] [PubMed]
- Gasmelseed, N.M.A.; Schmidt, M.; Magzoub, M.M.A.; Macharia, M.; Elmustafa, O.M.; Ototo, B.; Winkler, E.; Ruge, G.; Horstmann, R.D.; Meyer, C.G. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 2004, 23, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Javidnia, H.; Carson, N.; Awubwa, M.; Byaruhanga, R.; Mack, D.; Vaccani, J.-P. Connexin gene mutations among ugandan patients with nonsyndromic sensorineural hearing loss. Laryngoscope 2014, 124, E373–E376. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Lasisi, A.O.; Bademci, G.; Foster, J.; Blanton, S.; Tekin, M. Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: A report from Nigeria. Int. J. Pediatric Otorhinolaryngol. 2014, 78, 1870–1873. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Chobot-Rodd, J.; Castellanos, R.; Babcock, M.; Shanske, A.; Parikh, S.R.; Morrow, B.E.; Samanich, J. GJB2 mutation spectrum in 209 hearing impaired individuals of predominantly Caribbean Hispanic and African descent. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 611–618. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W Mutation Associated with Recessive Nonsyndromic Sensorineural Deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; De Kock, C.; Nembaware, V.; Quaye, O.Q.; Amedofu, G.K.K.; Awandare, G.; Wonkam, A. GJB2 and GJB6 mutations in non-syndromic childhood hearing impairment in Ghana. Front. Genet. 2019, 10, 841. [Google Scholar] [CrossRef]
- Ross, S.A.; Novak, Z.; Kumbla, R.A.; Zhang, K.; Fowler, K.B.; Boppana, S. GJB2 and GJB6 mutations in children with congenital cytomegalovirus infection. Pediatr. Res. 2007, 61, 687–691. [Google Scholar] [CrossRef]
- Shi, L.; Chen, J.; Li, J.; Wei, X.; Gao, X. Prevalence of GJB2 gene mutation in 330 cochlear implant patients in the Jiangsu province. J. Laryngol. Otol. 2016, 130, 902–906. [Google Scholar] [CrossRef]
- Rudman, J.R.; Kabahuma, R.I.; Bressler, S.E.; Feng, Y.; Blanton, S.H.; Yan, D.; Liu, X.-Z. The genetic basis of deafness in populations of African descent. J. Genet. Genom. 2017, 44, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, D.; Denoyelle, F.; Chauvin, P.; Garabédian, E.-N.; Couderc, R.; Odent, S.; Joannard, A.; Schmerber, S.; Delobel, B.; Leman, J.; et al. Large deletion of the GJB6 gene in deaf patients heterozygous for the GJB2 gene mutation: Genotypic and phenotypic analysis. Am. J. Med Genet. Part A 2004, 127A, 263–267. [Google Scholar] [CrossRef]
- Batissoco, A.C.; Abreu-Silva, R.S.; Braga, M.C.C.; Lezirovitz, K.; Della-Rosa, V.; Alfredo, T.; Otto, P.A.; Mingroni-Netto, R.C. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: Implications for diagnosis and genetic counseling. Ear Hear. 2009, 30, 1–7. [Google Scholar] [CrossRef]
- Dragomir, C.; Stan, A.; Stefanescu, D.T.; Savu, L.; Severin, E. Prenatal screening for the 35delG GJB2, del (GJB6-D13S1830), and del (GJB6-D13S1854) mutations in the Romanian population. Genet. Test. Mol. Biomark. 2011, 15, 749–753. [Google Scholar] [CrossRef]
- Bhalla, S.; Sharma, R.; Khandelwal, G.; Panda, N.K.; Khullar, M. Absence of GJB6 mutations in Indian patients with non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 356–359. [Google Scholar] [CrossRef]
- Naddafnia, H.; Noormohammadi, Z.; Irani, S.; Salahshoorifar, I. Frequency of GJB2 mutations, GJB6-D13S1830 and GJB6-D13S1854 deletions among patients with non-syndromic hearing loss from the central region of Iran. Mol. Genet. Genom. Med. 2019, 7, e00780. [Google Scholar]
- Amorini, M.; Romeo, P.; Bruno, R.; Galletti, F.; Di Bella, C.; Longo, P.; Briuglia, S.; Salpietro, C.; Rigoli, L. Prevalence of Deafness-Associated Connexin-26 (GJB2) and Connexin-30 (GJB6) Pathogenic Alleles in a Large Patient Cohort from Eastern Sicily. Ann. Hum. Genet. 2015, 79, 341–349. [Google Scholar] [CrossRef]
- Primignani, P.; Trotta, L.; Castorina, P.; Lalatta, F.; Sironi, F.; Radaelli, C.; Degiorgio, D.; Curcio, C.; Travi, M.; Ambrosetti, U.; et al. Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: Frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomark. 2009, 13, 209–217. [Google Scholar] [CrossRef]
- Lebeko, K.; Sloan-Heggen, C.M.; Noubiap, J.J.N.; Dandara, C.; Kolbe, D.L.; Ephraim, S.S.; Booth, K.T.; Azaiez, H.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 2016, 90, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Hertzano, R.; Elkon, R. High throughput gene expression analysis of the inner ear. Hear. Res. 2012, 288, 77–88. [Google Scholar] [CrossRef]
- Noubiap, J.-J.; Djomou, F.; Njock, R.; Toure, G.B.; Wonkam, A. Waardenburg syndrome in childhood deafness in Cameroon. S. Afr. J. Child Health 2014, 8, 3–5. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Level of Hearing Loss | N * | Percentage (%) |
|---|---|---|
| Severe I (71–80 dB) | 01 | 1.8 |
| Severe II (81–90 dB) | 02 | 3.7 |
| Profound I (91–100 dB) | 04 | 7.4 |
| Profound II (101–110 dB) | 13 | 24.1 |
| Profound III (111–119 dB) | 23 | 42.6 |
| Total (≥120 dB) | 11 | 20.4 |
| Total | 54 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tingang Wonkam, E.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.; F. Fokouo, J.V.; Wonkam, A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes 2019, 10, 844. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110844
Tingang Wonkam E, Chimusa E, Noubiap JJ, Adadey SM, F. Fokouo JV, Wonkam A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes. 2019; 10(11):844. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110844
Chicago/Turabian StyleTingang Wonkam, Edmond, Emile Chimusa, Jean Jacques Noubiap, Samuel Mawuli Adadey, Jean Valentin F. Fokouo, and Ambroise Wonkam. 2019. "GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon" Genes 10, no. 11: 844. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110844