Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethitical Approval
2.2. Genetic Analysis
2.3. Molecular Modeling
2.4. Plasmid Generation
2.5. Cell Culture
2.6. Immunocytochemistry and Confocal Microscopy
2.7. Statistics
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quinones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.L.; Roberts, R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Hayashi, T.; Nishi, H.; Kusaba, K.; Koga, Y.; Koga, Y.; Nonaka, I.; Kimura, A. Phenotypic expression of a novel desmin gene mutation: Hypertrophic cardiomyopathy followed by systemic myopathy. J. Hum. Genet. 2018, 63, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Draminska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 2007, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.E.; Veenstra-Knol, H.E.; van Essen, A.J.; van Ravenswaaij, C.M.; den Dunnen, W.F.; van den Wijngaard, A.; van Tintelen, J.P. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur. J. Med. Genet. 2007, 50, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschroder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Bar, H.; Mucke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [PubMed]
- Maerkens, A.; Kley, R.A.; Olive, M.; Theis, V.; van der Ven, P.F.; Reimann, J.; Milting, H.; Schreiner, A.; Uszkoreit, J.; Eisenacher, M.; et al. Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J. Proteom. 2013, 90, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Kreplak, L.; Bar, H. Severe myopathy mutations modify the nanomechanics of desmin intermediate filaments. J. Mol. Biol. 2009, 385, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef] [PubMed]
- Koster, S.; Weitz, D.A.; Goldman, R.D.; Aebi, U.; Herrmann, H. Intermediate filament mechanics in vitro and in the cell: From coiled coils to filaments, fibers and networks. Curr. Opin. Cell Biol. 2015, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Azzimato, V.; Genneback, N.; Tabish, A.M.; Buyandelger, B.; Knoll, R. Desmin, desminopathy and the complexity of genetics. J. Mol. Cell. Cardiol. 2016, 92, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- World Medical, A. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar]
- Chernyatina, A.A.; Nicolet, S.; Aebi, U.; Herrmann, H.; Strelkov, S.V. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 13620–13625. [Google Scholar] [CrossRef] [PubMed]
- Sarria, A.J.; Lieber, J.G.; Nordeen, S.K.; Evans, R.M. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J. Cell Sci. 1994, 107, 1593–1607. [Google Scholar] [PubMed]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Kedia, N.; Arhzaouy, K.; Pittman, S.K.; Sun, Y.; Batchelor, M.; Weihl, C.C.; Bieschke, J. Desmin forms toxic, seeding-competent amyloid aggregates that persist in muscle fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 16835–16840. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol. Genet. Genom. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- Arbustini, E.; Morbini, P.; Grasso, M.; Fasani, R.; Verga, L.; Bellini, O.; Dal Bello, B.; Campana, C.; Piccolo, G.; Febo, O.; et al. Restrictive cardiomyopathy, atrioventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J. Am. Coll. Cardiol. 1998, 31, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Bertini, E.; Bosman, C.; Ricci, E.; Servidei, S.; Boldrini, R.; Sabatelli, M.; Salviati, G. Neuromyopathy and restrictive cardiomyopathy with accumulation of intermediate filaments: A clinical, morphological and biochemical study. Acta Neuropathol. 1991, 81, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Zachara, E.; Bertini, E.; Lioy, E.; Boldrini, R.; Prati, P.L.; Bosman, C. Restrictive cardiomyopathy due to desmin accumulation in a family with evidence of autosomal dominant inheritance. G. Ital. Cardiol. 1997, 27, 436–442. [Google Scholar] [PubMed]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Ojrzynska, N.; Bilinska, Z.T.; Franaszczyk, M.; Ploski, R.; Grzybowski, J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol. Pol. 2017, 75, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudkova, A.; Kostareva, A.; Sjoberg, G.; Smolina, N.; Turalchuk, M.; Kuznetsova, I.; Rybakova, M.; Edstrom, L.; Shlyakhto, E.; Sejersen, T. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr. Cardiol. 2013, 34, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Qiang, G.; Jingmin, W.; Yanling, Y.; Xiru, W.; Yuwu, J. Clinical and genetic study in Chinese patients with Alexander disease. J. Child Neurol. 2008, 23, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Becane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Pinol-Ripoll, G.; Shatunov, A.; Cabello, A.; Larrode, P.; de la Puerta, I.; Pelegrin, J.; Ramos, F.J.; Olive, M.; Goldfarb, L.G. Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul. Disord. NMD 2009, 19, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.H.; Saric, T.; Mehrjardi, N.Z.; Hamad, S.; Morad, M. Acid-Sensitive Ion Channels Are Expressed in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Dev. 2019, 28, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Kredel, S.; Oswald, F.; Nienhaus, K.; Deuschle, K.; Rocker, C.; Wolff, M.; Heilker, R.; Nienhaus, G.U.; Wiedenmann, J. mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS ONE 2009, 4, e4391. [Google Scholar] [CrossRef] [PubMed]



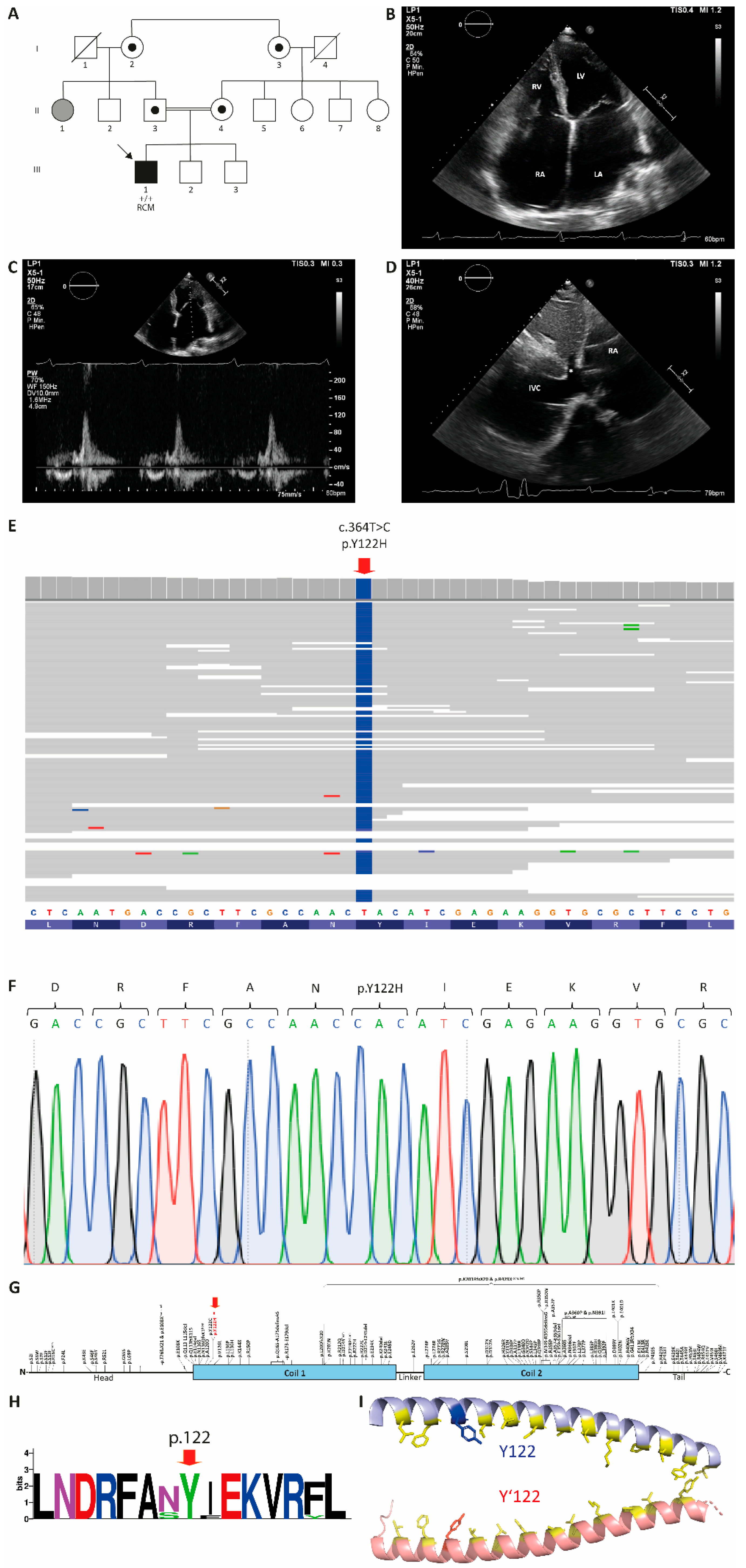{kind=link}
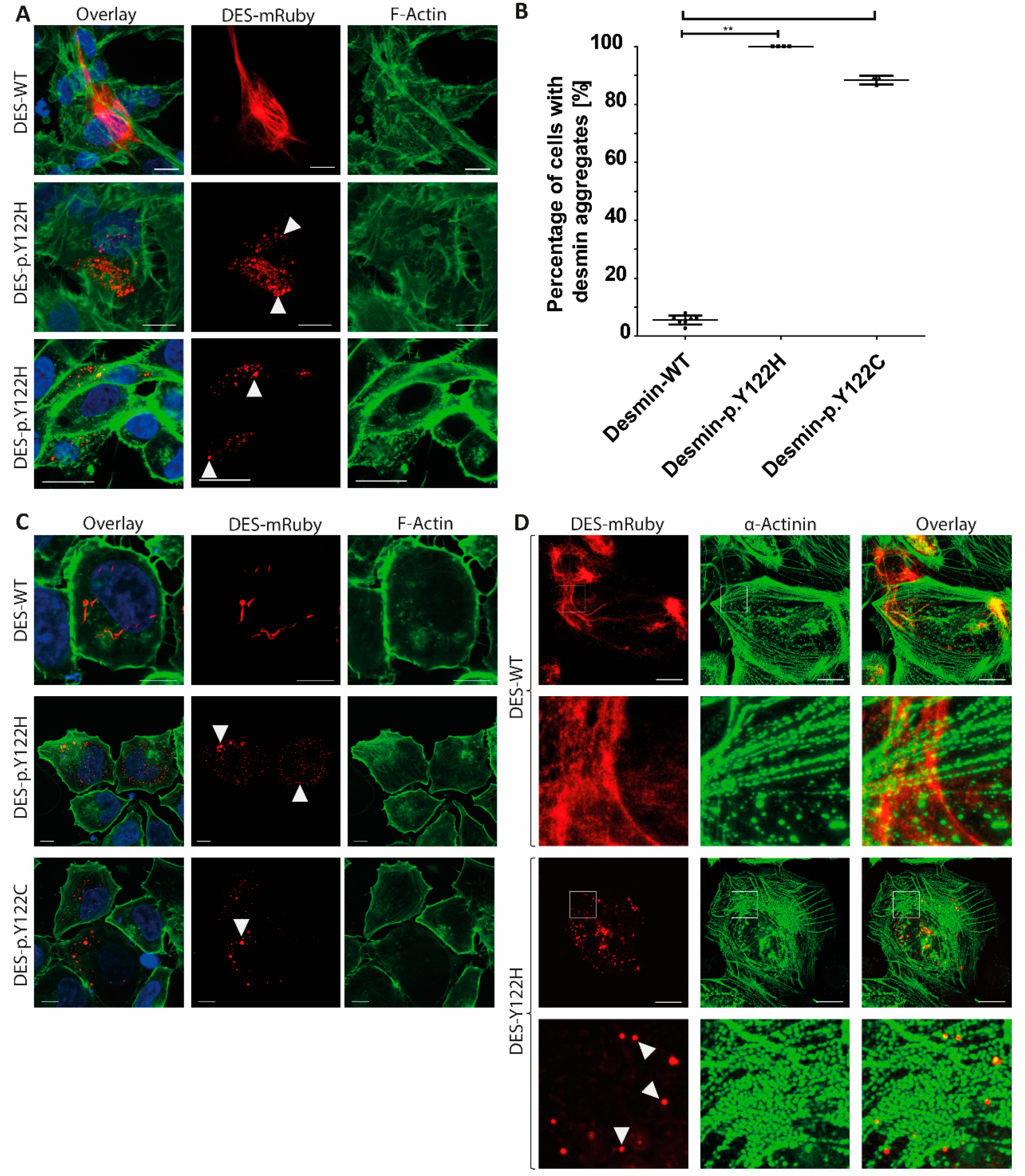{kind=link}
| PolyPhen-2 1 | Mutation Taster 2 | Provean 3 | Panther 4 | SNPs & GO 5 | SIFT |
|---|---|---|---|---|---|
| Probably damaging 0.999 | Disease causing 83 | Deleterious −4.228 | Probably damaging 750 | Disease-associated variant 0.613 | Deleterious |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110918
Brodehl A, Pour Hakimi SA, Stanasiuk C, Ratnavadivel S, Hendig D, Gaertner A, Gerull B, Gummert J, Paluszkiewicz L, Milting H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes. 2019; 10(11):918. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110918
Chicago/Turabian StyleBrodehl, Andreas, Seyed Ahmad Pour Hakimi, Caroline Stanasiuk, Sandra Ratnavadivel, Doris Hendig, Anna Gaertner, Brenda Gerull, Jan Gummert, Lech Paluszkiewicz, and Hendrik Milting. 2019. "Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect" Genes 10, no. 11: 918. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110918