Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis

 ,
,  and
and 
Abstract
:1. Introduction
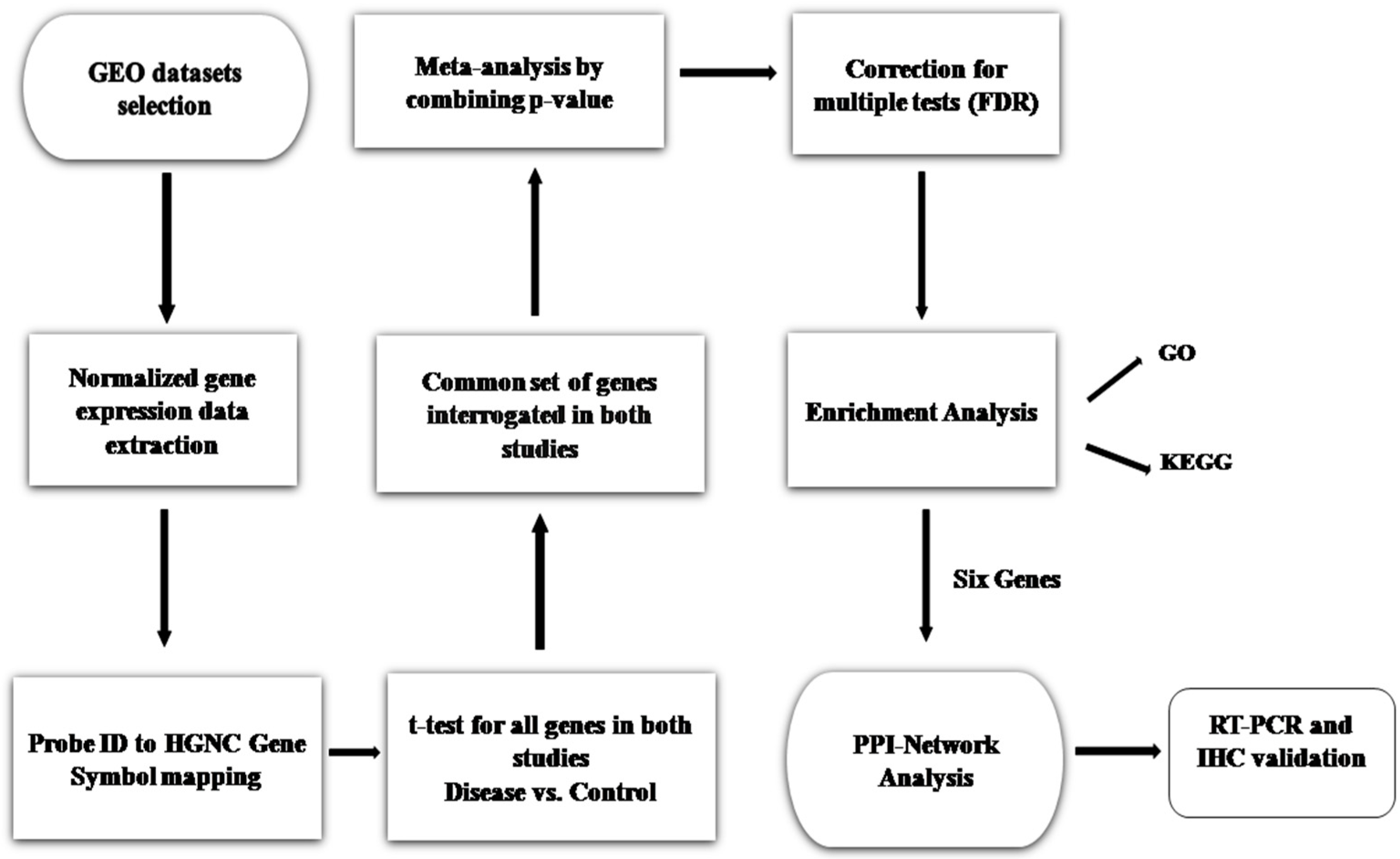
2. Materials and Methods
2.1. Sepsis-Associated Gene Expression Dataset Extraction
2.2. Meta-Analysis and DEGs Screening
2.3. Functional and Pathway Enrichment Analysis
2.4. PPI Network Construction and Analysis
2.5. Animal Model
2.6. Experimental Protocol
2.7. Semiquantitative RT-PCR
2.8. Immunohistochemistry for MPO
2.9. Statistical Analysis
3. Results
3.1. Sepsis-Associated Microarray Dataset Selection
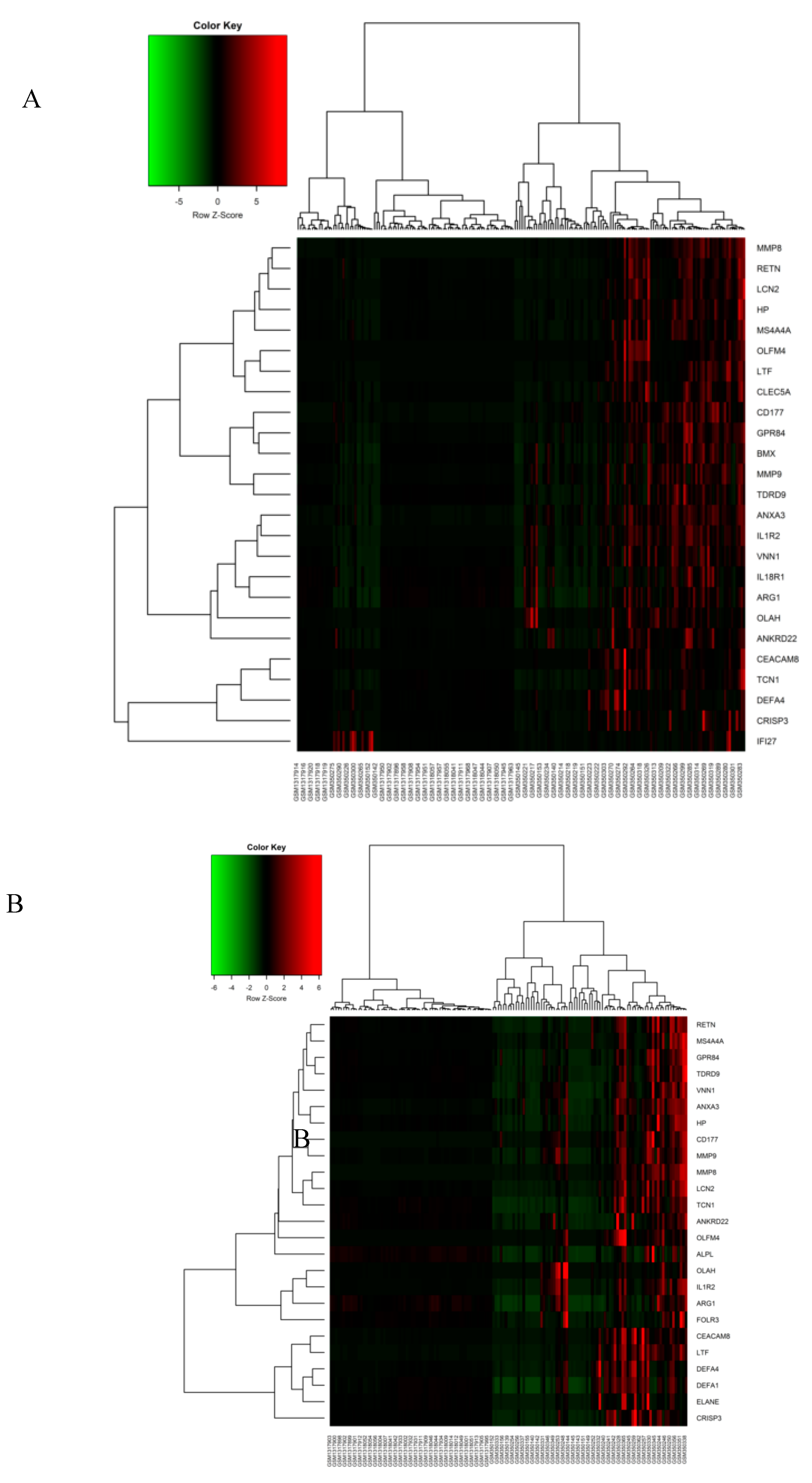
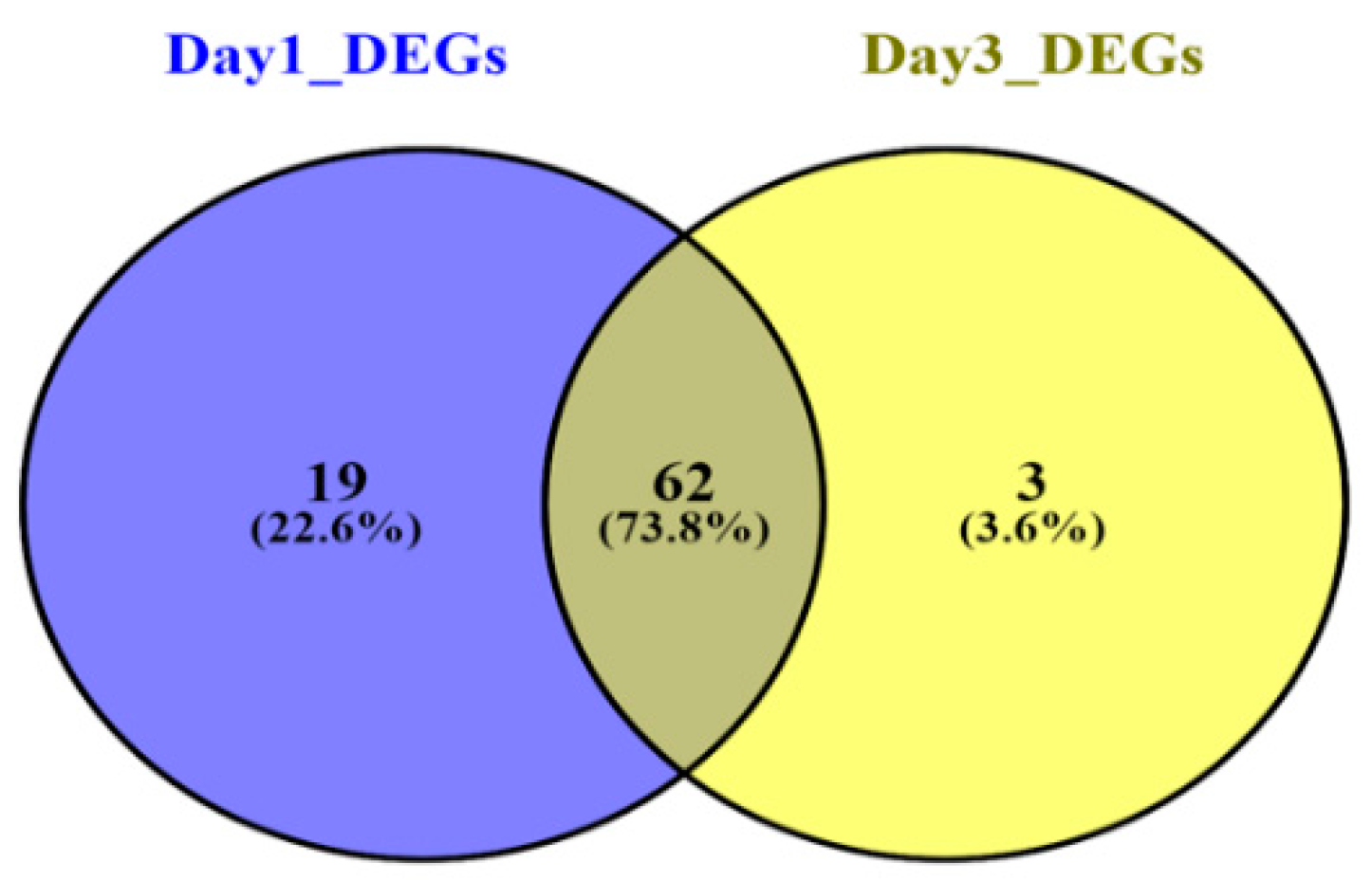
3.2. Meta-Analysis of Sepsis Datasets and DEGs Screening
3.3. Pathway and Functional Enrichment Analysis
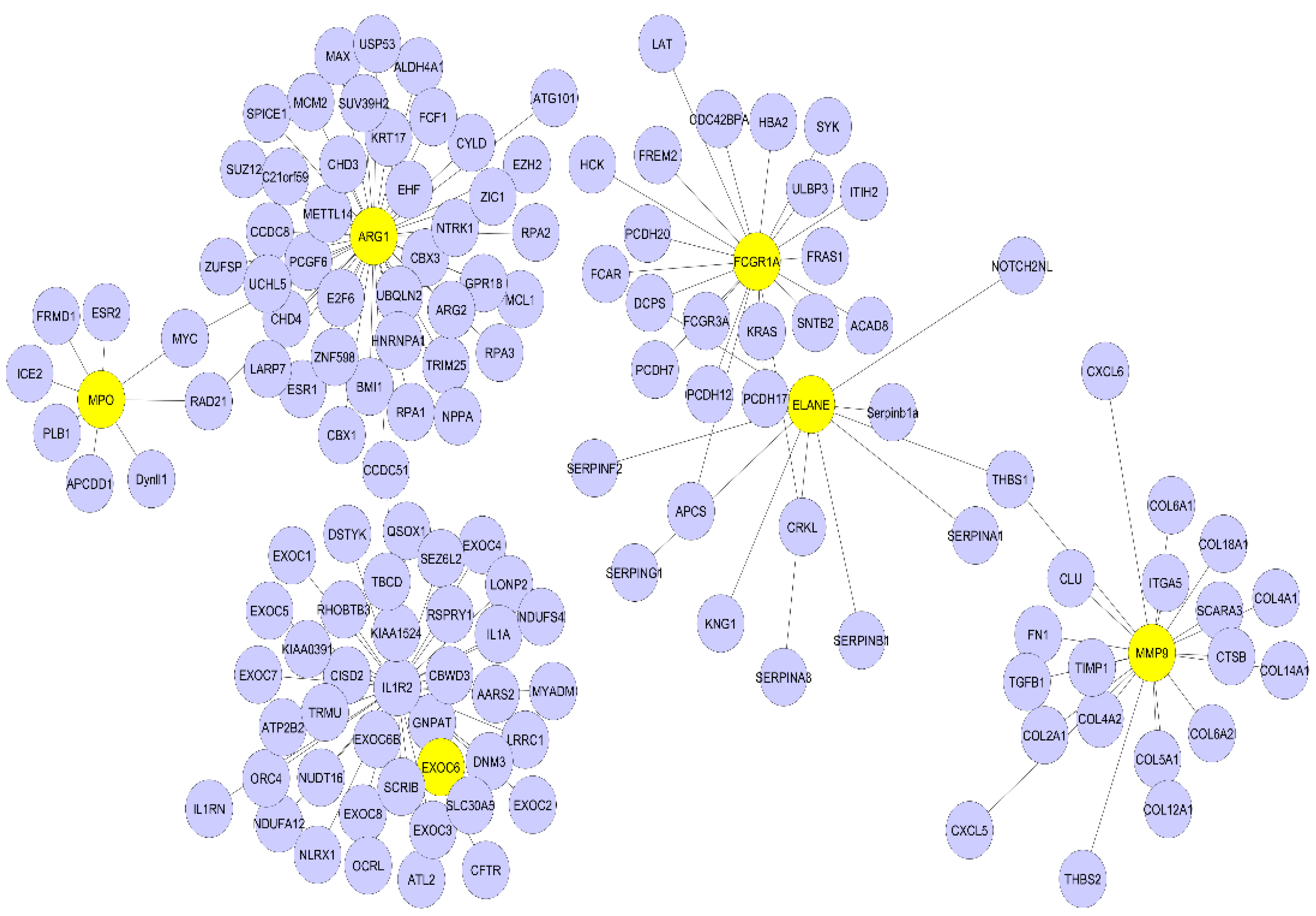
3.4. PPI Network Analysis
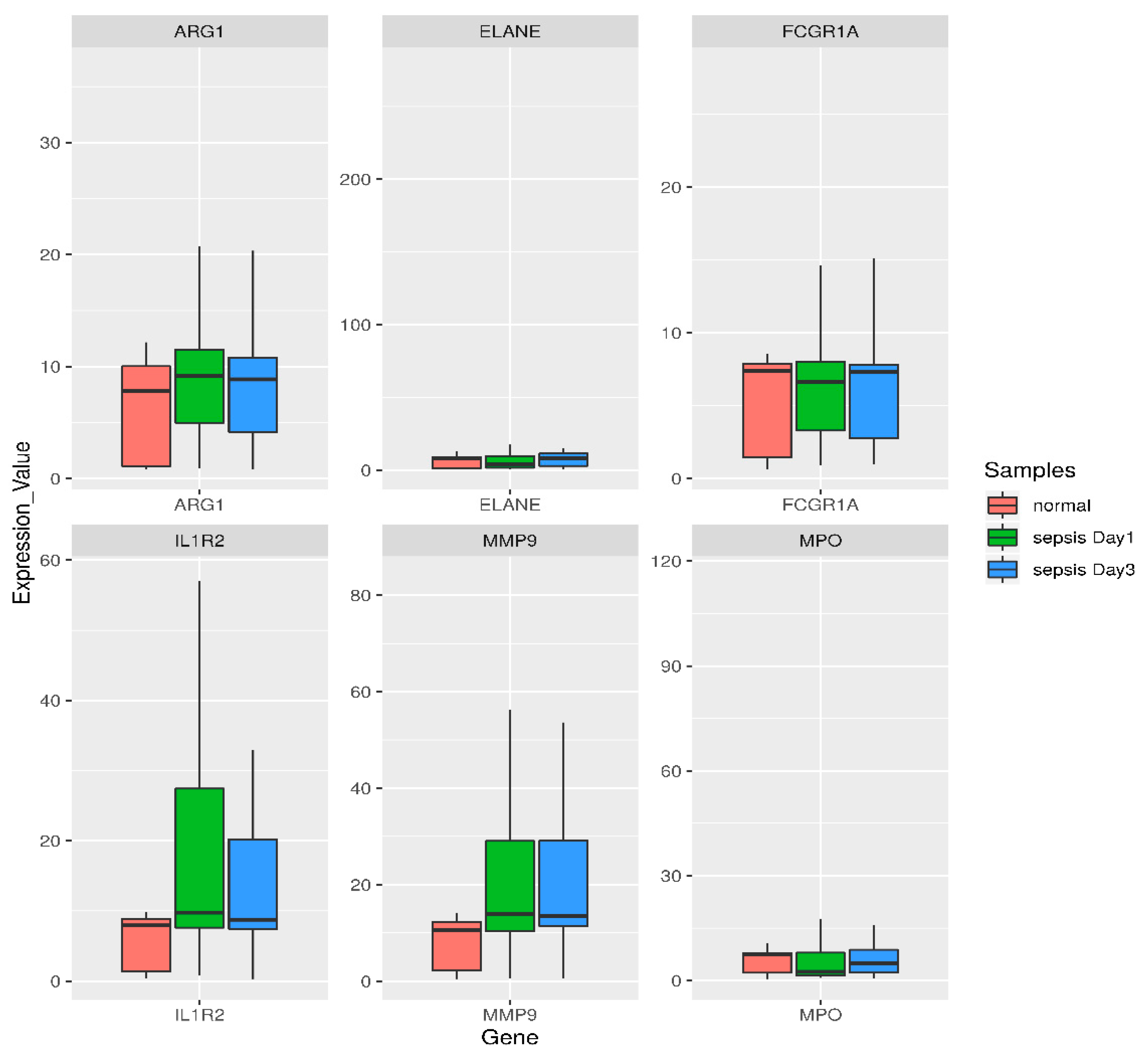
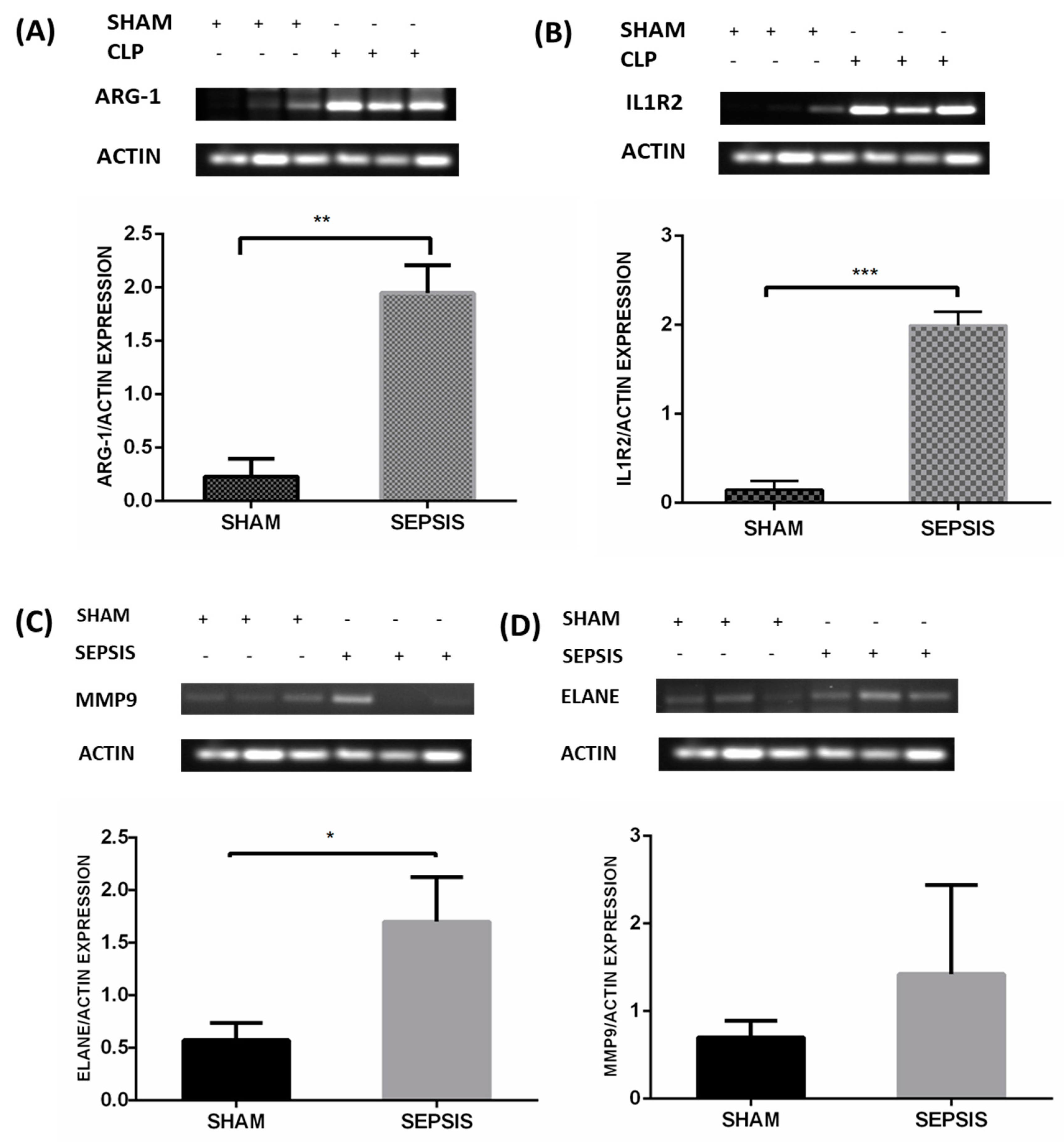
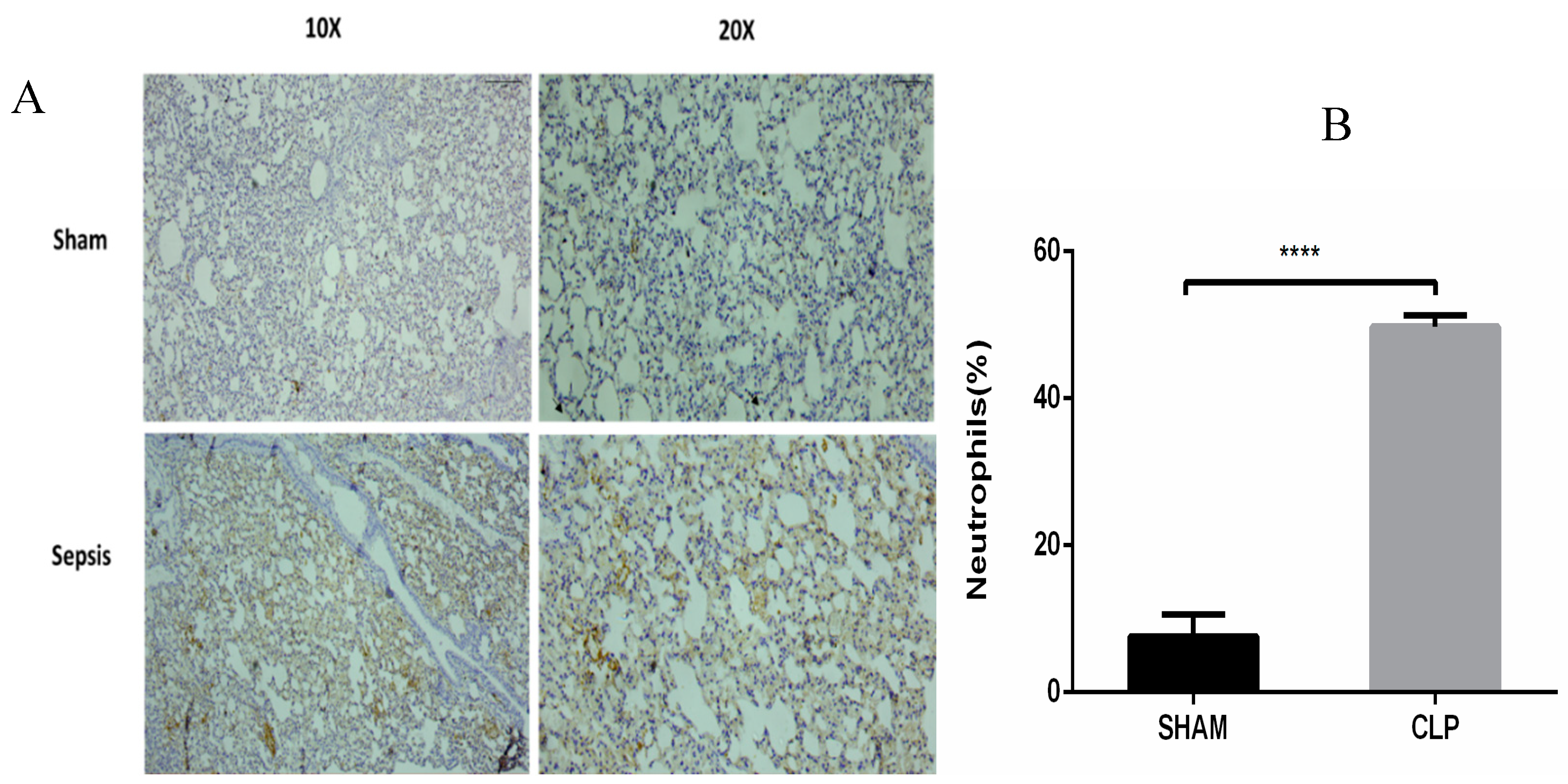
3.5. Semiquantitative RT-PCR Validation and Immunohistochemistry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Data and Materials
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G. Pathophysiology of Sepsis. Am. J. Pathol. 2007, 170, 1435–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ. 2016, 353, i1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, T.I.A.; Nielsen, G.G.; Andersen, P.K.; Teasdale, T.W. Genetic and Environmental Influences on Premature Death in Adult Adoptees. N. Engl. J. Med. 1988, 318, 727–732. [Google Scholar] [CrossRef]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key Issues in Conducting a Meta-Analysis of Gene Expression Microarray Datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.P.; McLean, A.S.; Dawes, I.W.; Huang, S.J.; Lin, R.C.Y. The Use of Gene-Expression Profiling to Identify Candidate Genes in Human Sepsis. Am. J. Respir. Crit. Care Med. 2007, 176, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.S.; Wei, C.; Yun, J.; Qian, L.X. Insights into the molecular mechanisms in sepsis with microarray technology. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2405–2412. [Google Scholar] [PubMed]
- Wang, Y.; Yin, X.; Yang, F. Comprehensive Analysis of Gene Expression Profiles of Sepsis-Induced Multiorgan Failure Identified Its Valuable Biomarkers. DNA Cell Biol. 2018, 37, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Slonim, D.K.; Yanai, I. Getting started in gene expression microarray analysis. PLoS Comput. Biol. 2009, 5, e1000543. [Google Scholar] [CrossRef]
- Elashoff, M.; Higgs, B.W.; Yolken, R.H.; Knable, M.B.; Weis, S.; Webster, M.J.; Barci, B.M.; Torrey, E.F. Meta-analysis of 12 genomic studies in bipolar disorder. J. Mol. Neurosci. 2007, 31, 221–243. [Google Scholar] [PubMed]
- Wong, H.R.; Cvijanovich, N.; Allen, G.L.; Lin, R.; Anas, N.; Meyer, K.; Freishtat, R.J.; Monaco, M.; Odoms, K.; Sakthivel, B.; et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit. Care Med. 2009, 37, 1558–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, G.P.; Tang, B.M.; Nalos, M.; Armstrong, N.J.; Huang, S.J.; Booth, D.R.; McLean, A.S. Identifying key regulatory genes in the whole blood of septic patients to monitor underlying immune dysfunctions. Shock 2013, 40, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics; Humana Press: New York, NY, USA, 2016; Volume 1418, pp. 93–110. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, C. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Schulz, K.F.; Simera, I.; Altman, D.G. Guidance for developers of health research reporting guidelines. PLoS Med. 2010, 7, e1000217. [Google Scholar] [CrossRef] [Green Version]
- Gray, K.A.; Seal, R.L.; Tweedie, S.; Wright, M.W.; Bruford, E.A. A review of the new HGNC gene family resource. Hum. Genomics 2016, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Berriz, G.F.; Roth, F.P. The Synergizer service for translating gene, protein and other biological identifiers. Bioinforma. Oxf. Engl. 2008, 24, 2272–2273. [Google Scholar] [CrossRef] [Green Version]
- Mudunuri, U.; Che, A.; Yi, M.; Stephens, R.M. bioDBnet: The biological database network. Bioinforma. Oxf. Engl. 2009, 25, 555–556. [Google Scholar] [CrossRef] [Green Version]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Mohammad, F.; Flight, R.M.; Harrison, B.J.; Petruska, J.C.; Rouchka, E.C. AbsIDconvert: An absolute approach for converting genetic identifiers at different granularities. BMC Bioinform. 2012, 13, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shriwash, N.; Singh, P.; Arora, S.; Ali, S.M.; Ali, S.; Dohare, R. Identification of differentially expressed genes in small and non-small cell lung cancer based on meta-analysis of mRNA. Heliyon 2019, 5, e01707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, S.H.; Haitsma, J.J.; Dos Santos, C.C.; Deng, Y.; Lai, P.F.; Slutsky, A.S.; Liles, W.C.; Stewart, D.J. Mesenchymal Stem Cells Reduce Inflammation while Enhancing Bacterial Clearance and Improving Survival in Sepsis. Am. J. Respir. Crit. Care Med. 2010, 182, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Tamizhselvi, R.; Manikandan, J.; Melendez, A.J.; Moochhala, S.M.; Bhatia, M. Substance P in polymicrobial sepsis: Molecular fingerprint of lung injury in preprotachykinin-A−/− mice. Mol. Med. Camb. Mass 2010, 16, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.L. On the Comparison of Several Mean Vaules: An Alternative Approach. Biometrika 1951, 38, 330. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers, 14th ed.; Revised and Enlarged; Oliver and Boyd: Edinburgh, Scotland, 1970. [Google Scholar]
- Haynes, W. Benjamini–Hochberg Method. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; p. 78. [Google Scholar]
- Da Wei Huang, B.T.S.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Panda, S.K.; Agarwal, B.; Behera, S.; Ali, S.M.; Pulse, M.E.; Solomkin, J.S.; Opal, S.M.; Bhandari, V.; Acharya, S. Novel Chitohexaose Analog Protects Young and Aged mice from CLP Induced Polymicrobial Sepsis. Sci. Rep. 2019, 9, 2904. [Google Scholar] [CrossRef]
- Varisco, B.M. The Pharmacology of Acute Lung Injury in Sepsis. Adv. Pharmacol. Sci. 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Marriott, H.M.; Ali, F.; Read, R.C.; Mitchell, T.J.; Whyte, M.K.B.; Dockrell, D.H. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004, 18, 1126–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcy, C.J.; Woodberry, T.; Davis, J.S.; Piera, K.A.; McNeil, Y.R.; Chen, Y.; Yeo, T.W.; Weinberg, J.B.; Anstey, N.M. Increased plasma arginase activity in human sepsis: Association with increased circulating neutrophils. Clin. Chem. Lab. Med. 2014, 52, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.A.; Joesting, J.J.; Freund, G.G. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain. Behav. Immun. 2013, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, Y.; Jiang, Y.; Gao, M.; Wang, W.; Wang, N.; Wang, K.; Zhang, H.; Chen, G.; Liu, K.; Liu, M.; et al. Interleukin-1 Receptor 2: A New Biomarker for Sepsis Diagnosis and Gram-Negative/Gram-Positive Bacterial Differentiation. Shock 2017, 47, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lalu, M.M.; Cena, J.; Chowdhury, R.; Lam, A.; Schulz, R. Matrix metalloproteinases contribute to endotoxin and interleukin-1 β induced vascular dysfunction. Br. J. Pharmacol. 2006, 149, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuibban, G.A.; Gong, J.H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Labarta, L.; Díaz, C.; Solé-Violán, J.; Blanquer, J.; Orbe, J.; Rodríguez, J.A.; Jiménez, A.; Borreguero-León, J.M.; et al. Matrix metalloproteinase-9, -10, and tissue inhibitor of matrix metalloproteinases-1 blood levels as biomarkers of severity and mortality in sepsis. Crit. Care 2009, 13, R158. [Google Scholar] [CrossRef] [Green Version]
- Vandooren, J.; Swinnen, W.; Ugarte-Berzal, E.; Boon, L.; Dorst, D.; Martens, E.; Opdenakker, G. Endotoxemia shifts neutrophils with TIMP-free gelatinase B/MMP-9 from bone marrow to the periphery and induces systematic upregulation of TIMP-1. Haematologica 2017, 102, 1671–1682. [Google Scholar] [CrossRef] [Green Version]
- Van den Steen, P.E.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and Molecular Biology of Gelatinase B or Matrix Metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 2002, 37, 375–536. [Google Scholar] [CrossRef]
- Rasooli, A.; Ghafari, E.; Saedi, H.; Miri, S. Expression changes of CD177 and MPO as novel biomarkers in lung tissue of CLP model rats. Turk. J. Med. Sci. 2018, 48, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.; Ode, Y.; Zhou, M.; Ochani, M.; Holodick, N.E.; Rothstein, T.L.; Wang, P. B-1a cells protect mice from sepsis-induced acute lung injury. Mol. Med. 2018, 24, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, K.; Horie, O.; Yoshioka, A.; Matsuki, S.; Tenjin, T.; Tsukamura, Y.; Yoneda, M.; Shibata, K.; Koike, Y.; Nomura, T.; et al. Association between the neutrophil myeloperoxidase index and subsets of bacterial infections: MXPI IN BACTERIAL INFECTION. Int. J. Lab. Hematol. 2010, 32, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxid. Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ebihara, I.; Shimada, N.; Shoji, H.; Koide, H. Modulation of Plasma Metalloproteinase-9 Concentrations and Peripheral Blood Monocyte mRNA Levels in Patients with Septic Shock: Effect of Fiber-Immobilized Polymyxin B Treatment. Am. J. Med. Sci. 1998, 316, 355–360. [Google Scholar] [PubMed]
- Maitra, S.R.; Bhaduri, S.; Valane, P.D.; Tervahartiala, T.; Sorsa, T.; Ramamurthy, N. Inhibition of matrix metalloproteinases by chemically modified tetracyclines in sepsis. Shock 2003, 20, 280–285. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Tsalik, E.L.; Langley, R.J.; Dinwiddie, D.L.; Miller, N.A.; Yoo, B.; van Velkinburgh, J.C.; Smith, L.D.; Thiffault, I.; Jaehne, A.K.; Valente, A.M.; et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med. 2014, 6, 111. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Hankey Giblin, P. Insights into Macrophage Heterogeneity and Cytokine-Induced Neuroinflammation in Major Depressive Disorder. Pharmaceuticals 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession Number | Disease | Sample | Sample Source | Platform | |
|---|---|---|---|---|---|
| Day1 | Day3 | ||||
| GSE13904 | Sepsis | n = 99 | n = 59 | Blood | Affymetrix Human Genome U 133 Plus 2.0 Array |
| GSE54514 | Sepsis | n = 35 | n = 38 | Blood | IlluminaHumanHT-12 V3.0 Expression BeadChip |
| Sepsis Day1 | Sepsis Day3 | ||||
|---|---|---|---|---|---|
| Gene | BH-p-Vaule | Fold Change | Gene | BH-p-Vaule | Fold Change |
| MMP8 | 3.03 × 10−12 | 90.83269343 | MMP8 | 1.25 × 10−6 | 53.11011783 |
| OLFM4 | 7.66 × 10−7 | 54.94062655 | OLFM4 | 0.000177074 | 33.23986192 |
| CD177 | 1.73 × 10−15 | 27.69570432 | CD177 | 2.68 × 10−7 | 24.4262987 |
| CEACAM8 | 2.01 × 10−6 | 17.21435595 | CEACAM8 | 1.54 × 10−6 | 18.71820844 |
| LTF | 3.79 × 10−11 | 14.76500984 | LTF | 1.35 × 10−7 | 14.33448059 |
| LCN2 | 5.64 × 10−9 | 12.69709454 | MMP9 | 4.75 × 10−10 | 13.37223858 |
| OLAH | 3.36 × 10−9 | 12.50259397 | OLAH | 0.000755255 | 11.87923585 |
| MMP9 | 6.75 × 10−17 | 11.6185382 | LCN2 | 8.36 × 10−7 | 10.78809213 |
| ANXA3 | 1.29 × 10−21 | 10.82040432 | DEFA4 | 3.74 × 10−8 | 10.37992756 |
| IL1R2 | 2.97 × 10−19 | 10.5634416 | IL1R2 | 2.86 × 10−8 | 8.994229615 |
| RETN | 4.97 × 10−12 | 10.42790653 | ANXA3 | 5.44 × 10−10 | 8.817467871 |
| HP | 8.36 × 10−13 | 10.0624041 | DEFA1 | 2.46 × 10−12 | 7.365054135 |
| GPR48 | 4.23 × 10−15 | 8.581561273 | RETN | 9.41 × 10−7 | 6.628552774 |
| ANKRD22 | 2.59 × 10−14 | 7.460137951 | HP | 9.96 × 10−7 | 6.35862726 |
| CLEC5A | 2.76 × 10−13 | 6.669614066 | MS4A4A | 4.37 × 10−5 | 6.16777804 |
| DEFA4 | 0.000155626 | 6.278035491 | ELANE | 9.42 × 10−5 | 5.65341365 |
| MS4A4A | 6.31 × 10−12 | 5.989831378 | VNN1 | 3.55 × 10−8 | 5.537816869 |
| VNN1 | 3.55 × 10−18 | 5.855759515 | GPR84 | 1.64 × 10−6 | 5.069264307 |
| TCN1 | 3.20 × 10−10 | 5.533269805 | TCN1 | 2.72 × 10−8 | 4.837923713 |
| GO ID | GO Term | No. of Genes | p-Vaule |
| Biological Process | |||
| GO:0045087 | innate immune response | 17 | 8.47 × 10−11 |
| GO:0006955 | immune response | 12 | 3.26 × 10−6 |
| GO:0042742 | defense response to bacterium | 8 | 4.49 × 10−6 |
| Molecular Functions | |||
| GO:0004252 | serine − type endopeptidase activity | 8 | 1.26 × 10−4 |
| GO:0008201 | heparin binding | 5 | 0.005346 |
| GO:0004869 | cysteine − type endopeptidase inhibitor activity | 3 | 0.009713 |
| Cellular Component | |||
| GO:0005615 | extracellular space | 27 | 2.49 × 10−11 |
| GO:0070062 | extracellular exosome | 38 | 2.57 × 10−11 |
| GO:0005576 | extracellular region | 26 | 6.39 × 10−9 |
| KEGG ID | KEGG Pathway | No. of Genes | p-Vaule |
| hsa05202 | Transcriptional misregulation in cancer | 6 | 9.47 × 10−4 |
| hsa05150 | Staphylococcus aureus infection | 4 | 0.001906 |
| hsa05134 | Legionellosis | 3 | 0.025598 |
| hsa05321 | Inflammatory bowel disease (IBD) | 3 | 0.035048 |
| hsa04610 | Complement and coagulation cascades | 3 | 0.040208 |
| hsa05146 | Amoebiasis | 3 | 0.085888 |
| GO ID | GO Term | No. of Genes | p-Vaule |
| Biological Process | |||
| GO:0045087 | Innate immune response | 14 | 3.65 × 10−9 |
| GO:0050832 | Defense response to fungus | 5 | 2.60 × 10−6 |
| GO:0042742 | Defense response to bacterium | 7 | 1.40 × 10−5 |
| Molecular Functions | |||
| GO:0004252 | Serine − type endopeptidase activity | 8 | 2.29 × 10−5 |
| GO:0008201 | Heparin binding | 5 | 0.002077 |
| GO:0008233 | Peptidase activity | 4 | 0.003483 |
| Cellular Component | |||
| GO:0070062 | Extracellular exosome | 35 | 8.32 × 10−13 |
| GO:0005615 | Extracellular space | 25 | 3.64 × 10−12 |
| GO:0005576 | Extracellular region | 21 | 1.83 × 10−7 |
| KEGG ID | KEGG Pathway | No. of Genes | p-Vaule |
| hsa05202 | Transcriptional misregulation in cancer | 5 | 0.001750917 |
| hsa05146 | Amoebiasis | 3 | 0.044114518 |
| hsa05322 | Systemic lupus erythematosus | 3 | 0.066980623 |
| hsa00052 | Galactose metabolism | 2 | 0.091415842 |
| hsa00051 | Fructose and mannose metabolism | 2 | 0.097217685 |
| Gene | Node Degree | Betweenness | Closeness |
|---|---|---|---|
| ARG1 | 43 | 0.78 | 0.49 |
| IL1R2 | 40 | 0.68 | 0.44 |
| FCGR1A | 21 | 0.63 | 0.34 |
| MMP9 | 20 | 0.61 | 0.33 |
| ELANE | 10 | 0.65 | 0.38 |
| MPO | 08 | 0.12 | 0.27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Singh, P.; Sharma, A.; Arora, S.; Shriwash, N.; Rahmani, A.H.; Almatroodi, S.A.; Manda, K.; Dohare, R.; Syed, M.A. Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis. Genes 2019, 10, 1005. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121005
Ahmad S, Singh P, Sharma A, Arora S, Shriwash N, Rahmani AH, Almatroodi SA, Manda K, Dohare R, Syed MA. Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis. Genes. 2019; 10(12):1005. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121005
Chicago/Turabian StyleAhmad, Shaniya, Prithvi Singh, Archana Sharma, Shweta Arora, Nitesh Shriwash, Arshad Husain Rahmani, Saleh A. Almatroodi, Kailash Manda, Ravins Dohare, and Mansoor Ali Syed. 2019. "Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis" Genes 10, no. 12: 1005. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121005