Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Animal Experiments
2.3. Immunofluorescence
2.4. In Vitro Experiment in Endothelial Cells
2.5. Western Blotting
2.6. DPP-4 Activity Detection
2.7. RNA Isolation and qPCR
2.8. RNA Extraction and microRNA Array Analysis
2.9. RNA Isolation and qPCR
2.10. Transfection
2.11. Statistical Analysis
3. Results
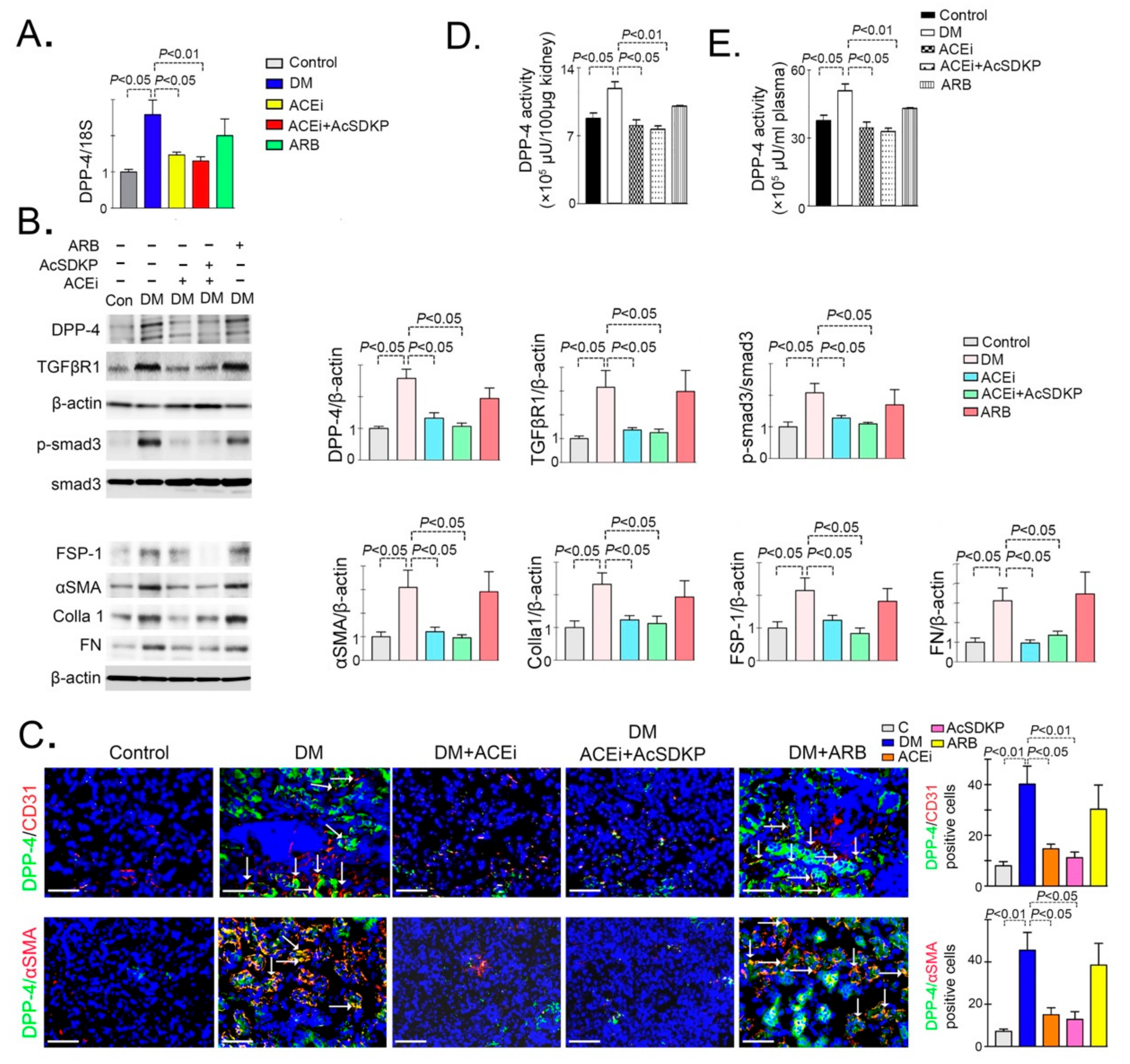
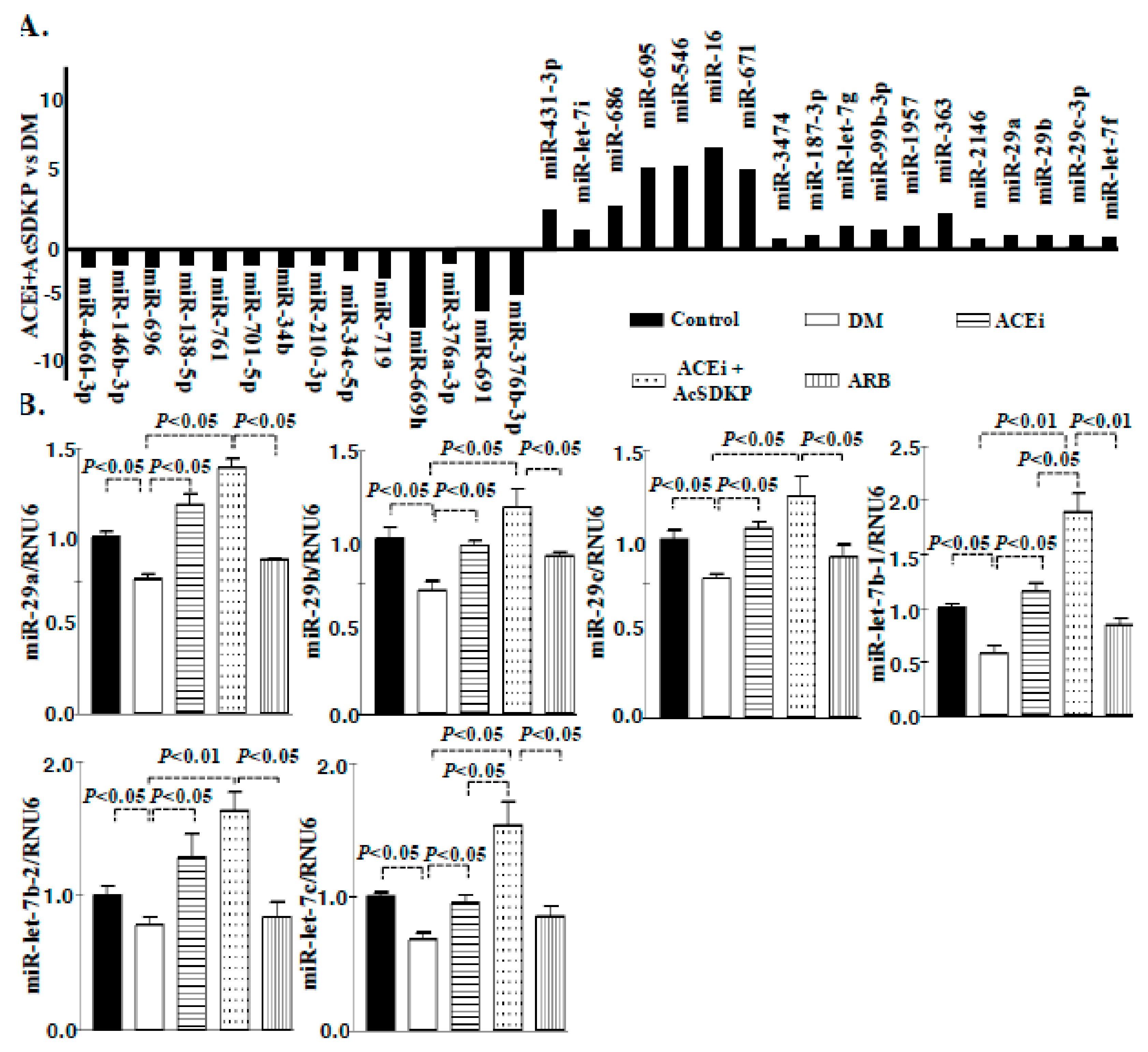
3.1. Inhibition of ACE Suppresses DPP-4 and Associated TGFβ Signaling in Diabetic Kidneys
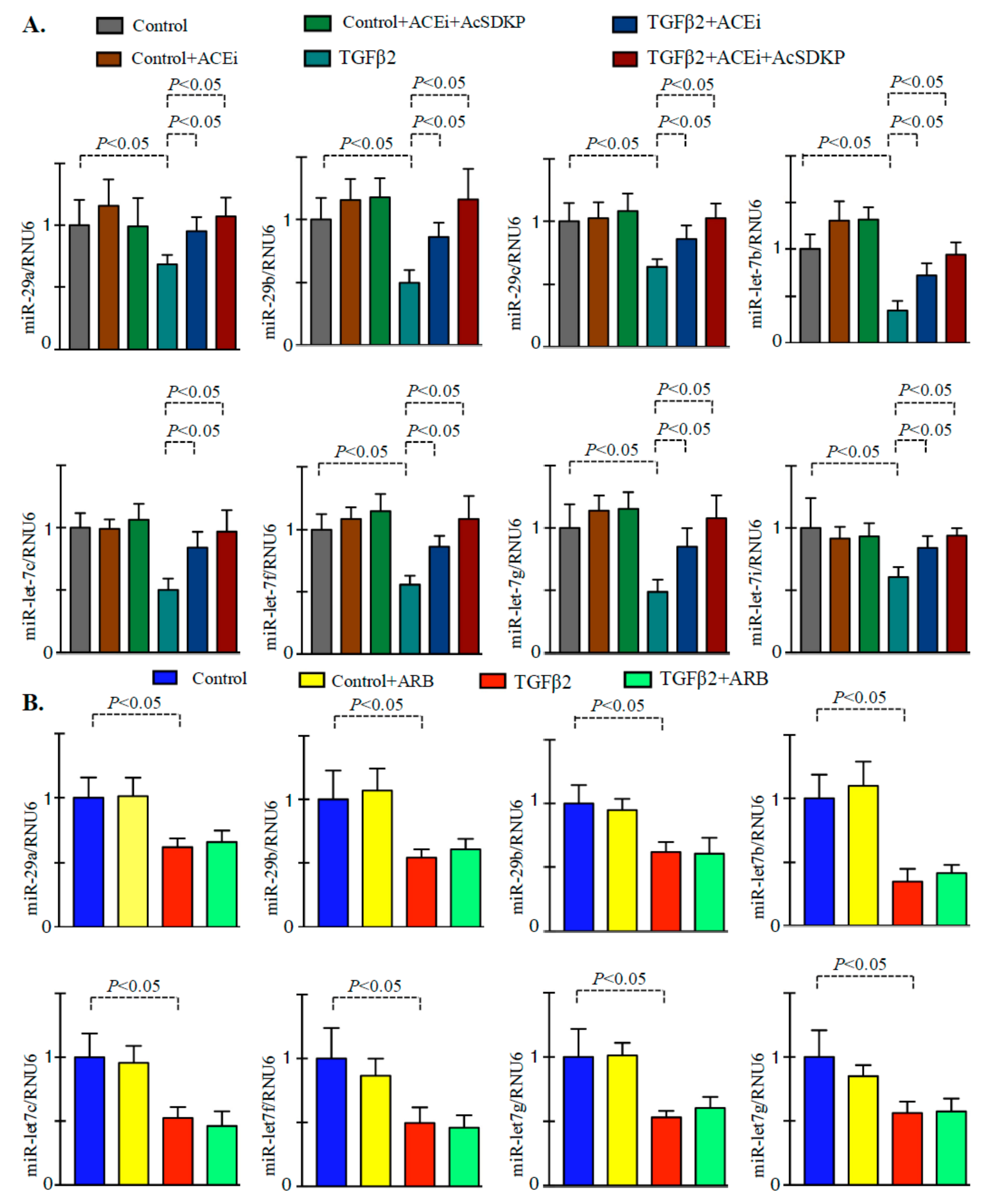
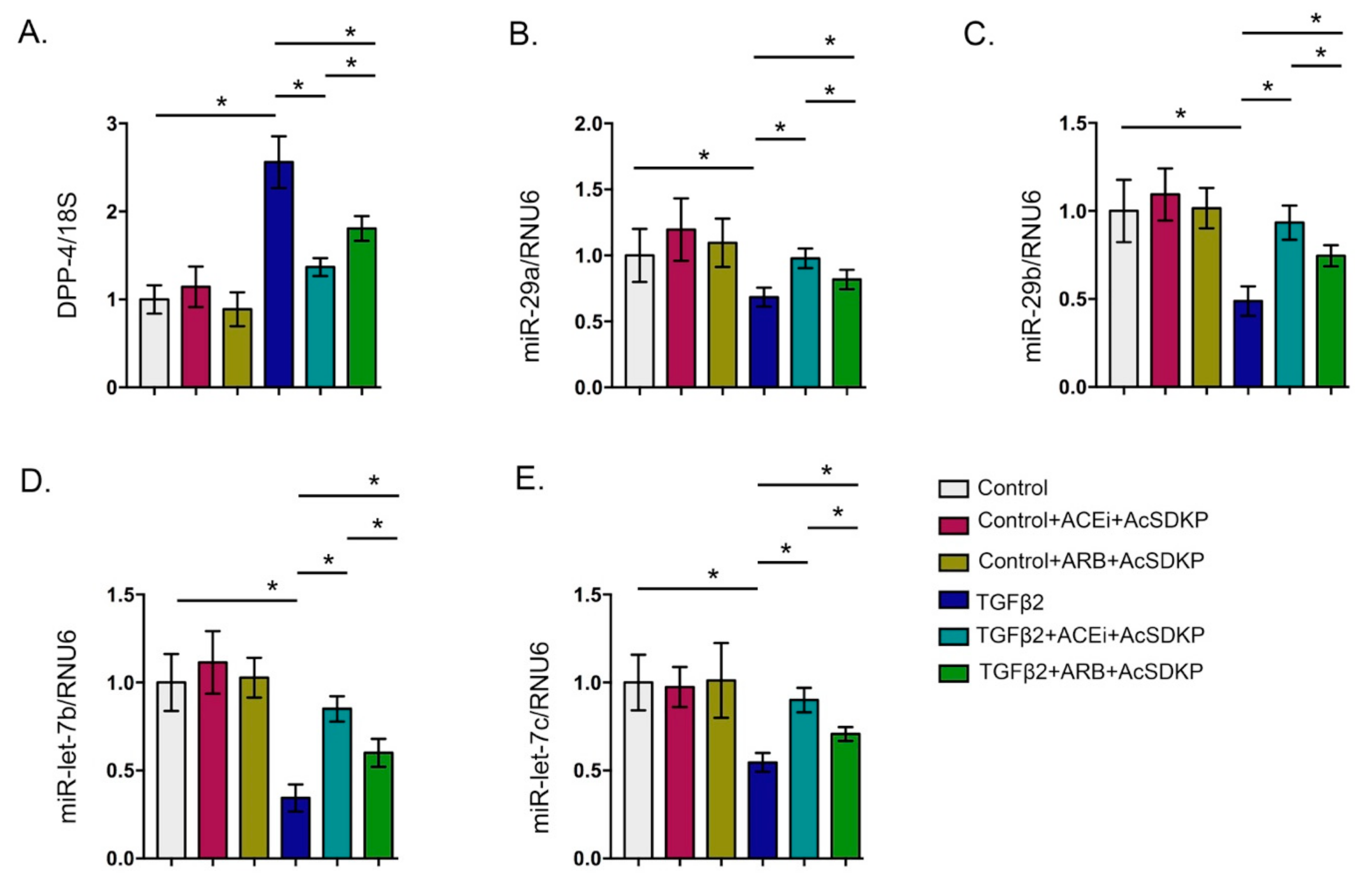
3.2. Inhibition of ACE Inhibits DPP-4 Level and Associated TGFβ Signaling in Endothelial Cells
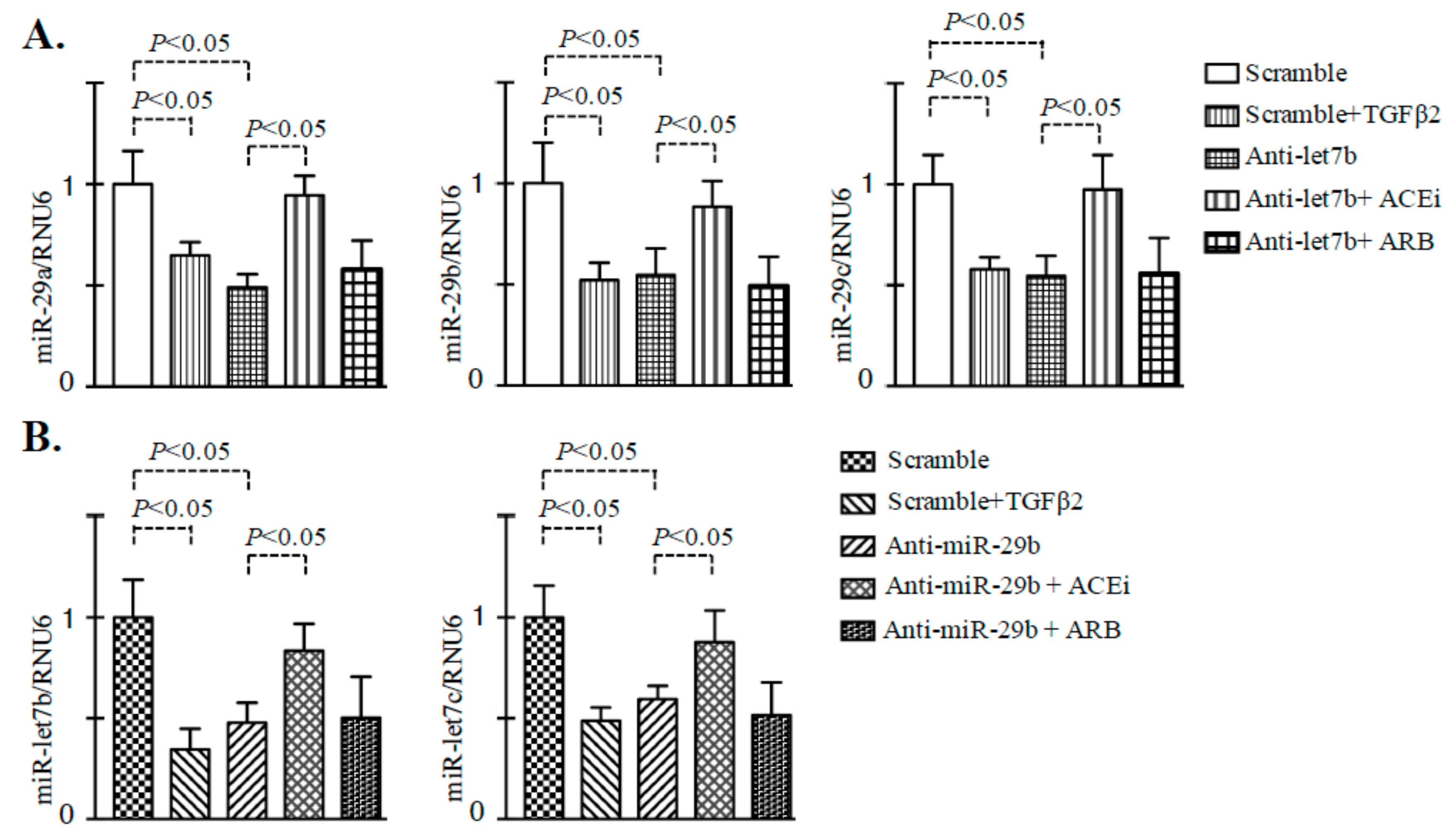
3.3. Inhibition of ACE Restored the TGFβ2-Associated Disruption of Cross-Talk Regulation between miR-29 and miR-let-7 Family Members in Endothelial Cells
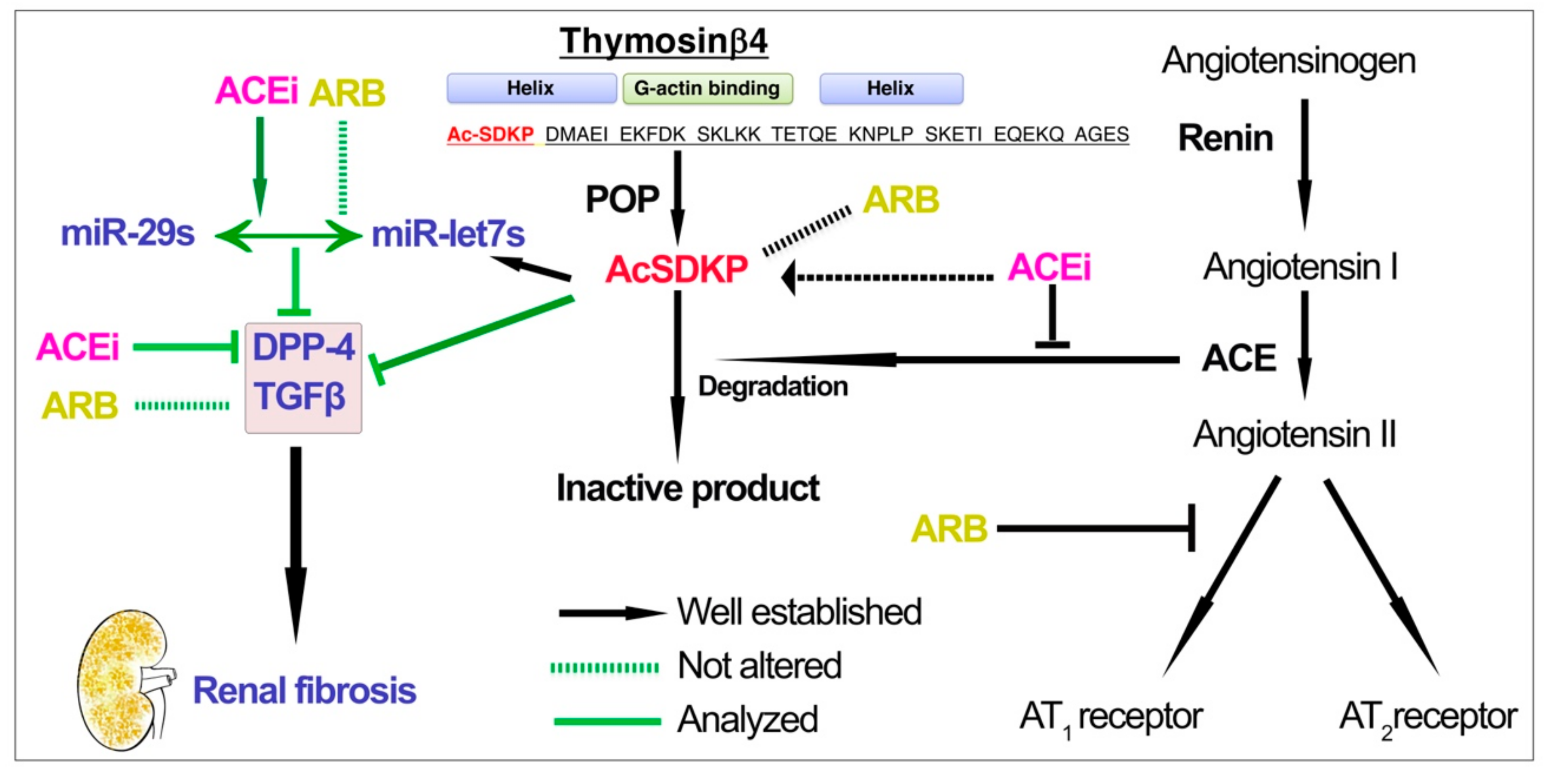
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Held, P.J.; Port, F.K.; Webb, R.L.; Wolfe, R.A.; Garcia, J.R.; Blagg, C.R.; Agodoa, L.Y. The united states renal data system’s 1991 annual data report: An introduction. Am. J. Kidney Dis. 1991, 18, 1–16. [Google Scholar]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Hedayat, F.A.; Kanasaki, K.; Goodwin, J.E. Microrna crosstalk influences epithelial-to-mesenchymal, endothelial-to-mesenchymal, and macrophage-to-mesenchymal transitions in the kidney. Front. Pharmacol. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, J.; Viswanathan, V. Management of diabetic hypertensives. Indian J. Endocrinol. Metab. 2011, 15 Suppl 4, S374–379. [Google Scholar]
- Hsu, F.Y.; Lin, F.J.; Ou, H.T.; Huang, S.H.; Wang, C.C. Renoprotective effect of angiotensin-converting enzyme inhibitors and angiotensin ii receptor blockers in diabetic patients with proteinuria. Kidney Blood Press Res. 2017, 42, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Mavridis, D.; Navarese, E.; Craig, J.C.; Tonelli, M.; Salanti, G.; Wiebe, N.; Ruospo, M.; Wheeler, D.C.; Strippoli, G.F. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: A network meta-analysis. Lancet 2015, 385, 2047–2056. [Google Scholar] [CrossRef]
- Gu, J.; Yang, M.; Qi, N.; Mei, S.; Chen, J.; Song, S.; Jing, Y.; Chen, M.; He, L.; Sun, L.; et al. Olmesartan prevents microalbuminuria in db/db diabetic mice through inhibition of angiotensin ii/p38/sirt1-induced podocyte apoptosis. Kidney Blood Press Res. 2016, 41, 848–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laverman, G.D.; Remuzzi, G.; Ruggenenti, P. Ace inhibition versus angiotensin receptor blockade: Which is better for renal and cardiovascular protection? J. Am. Soc. Nephrol. 2004, 15 Suppl 1, S64–70. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Kanasaki, M.; Srivastava, S.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Shi, S.; Kanasaki, K.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Von Websky, K.; Reichetzeder, C.; Hocher, B. Physiology and pathophysiology of incretins in the kidney. Curr. Opin. Nephrol. Hypertens. 2014, 23, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated dpp-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.; Cheng, Y.; Li, X.; He, M.; Zhu, J.; Han, H.; Wei, G.; Kong, H.; Xie, W.; et al. Glucagon-like peptide-1 mediates the protective effect of the dipeptidyl peptidase iv inhibitor on renal fibrosis via reducing the phenotypic conversion of renal microvascular cells in monocrotaline-treated rats. Biomed. Res. Int. 2018, 2018, 1864107. [Google Scholar]
- Uchida, T.; Oda, T.; Matsubara, H.; Watanabe, A.; Takechi, H.; Oshima, N.; Sakurai, Y.; Kumagai, H. Renoprotective effects of a dipeptidyl peptidase 4 inhibitor in a mouse model of progressive renal fibrosis. Ren. Fail 2017, 39, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.S.; Kim, J.E.; Lee, M.H.; Song, H.K.; Kang, Y.S.; Lee, M.J.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Chung, Y.Y.; et al. Dipeptidyl peptidase iv inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction. Lab Invest. 2014, 94, 598–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of dpp-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanasaki, M.; Nagai, T.; Kitada, M.; Koya, D.; Kanasaki, K. Elevation of the anti-fibrotic peptide n-acetyl-seryl-aspartyl-lysyl-proline: A blood pressure-independent beneficial effect of angiotensin i-converting enzyme inhibitors. Fibrogen. Tissue Rep. 2011, 4, 25. [Google Scholar]
- Kanasaki, K.; Koya, D.; Sugimoto, T.; Isono, M.; Kashiwagi, A.; Haneda, M. N-acetyl-seryl-aspartyl-lysyl-proline inhibits tgf-beta-mediated plasminogen activator inhibitor-1 expression via inhibition of smad pathway in human mesangial cells. J. Am. Soc. Nephrol. JASN 2003, 14, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Kanasaki, K.; Isono, M.; Sato, H.; Omata, M.; Sugimoto, T.; Araki, S.; Isshiki, K.; Kashiwagi, A.; Haneda, M.; et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes 2005, 54, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Omata, M.; Taniguchi, H.; Koya, D.; Kanasaki, K.; Sho, R.; Kato, Y.; Kojima, R.; Haneda, M.; Inomata, N. N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in wky rats with established anti-glomerular basement membrane nephritis. J. Am. Soc. Nephrol. JASN 2006, 17, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.; Shi, S.; Nagai, T.; Kanasaki, M.; Kitada, M.; Srivastava, S.P.; Haneda, M.; Kanasaki, K.; Koya, D. Oral administration of n-acetyl-seryl-aspartyl-lysyl-proline ameliorates kidney disease in both type 1 and type 2 diabetic mice via a therapeutic regimen. Biomed. Res. Int. 2016, 2016, 9172157. [Google Scholar] [CrossRef]
- Pandey, A.K.; Verma, G.; Vig, S.; Srivastava, S.; Srivastava, A.K.; Datta, M. Mir-29a levels are elevated in the db/db mice liver and its overexpression leads to attenuation of insulin action on pepck gene expression in hepg2 cells. Mol. Cell Endocrinol. 2011, 332, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Pandey, A.K.; Srivastava, S.; Srivastava, A.K.; Datta, M. Comprehensive mirnome and in silico analyses identify the wnt signaling pathway to be altered in the diabetic liver. Mol. Biosyst. 2011, 7, 3234–3244. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.R.D.; Domingueti, C.P. Micrornas: New biomarkers and promising therapeutic targets for diabetic kidney disease. J. Bras. Nefrol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorente-Cebrian, S.; Gonzalez-Muniesa, P.; Milagro, F.I.; Martinez, J.A. Micrornas and other non-coding rnas in adipose tissue and obesity: Emerging roles as biomarkers and therapeutic targets. Clin. Sci. (Lond) 2019, 133, 23–40. [Google Scholar] [CrossRef]
- Dehaini, H.; Awada, H.; El-Yazbi, A.; Zouein, F.A.; Issa, K.; Eid, A.A.; Ibrahim, M.; Badran, A.; Baydoun, E.; Pintus, G.; et al. Micrornas as potential pharmaco-targets in ischemia-reperfusion injury compounded by diabetes. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Chen, Z.; Zhou, Z.; Li, Y.; Wang, Y.; Zhou, Z.; Lu, H.; Sun, C.; Chu, X. High-throughput sequencing of small rnas and analysis of differentially expressed micrornas associated with high-fat diet-induced hepatic insulin resistance in mice. Genes Nutr. 2019, 14, 6. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Shi, S.; Kanasaki, M.; Nagai, T.; Kitada, M.; He, J.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; Koya, D. Effect of antifibrotic micrornas crosstalk on the action of n-acetyl-seryl-aspartyl-lysyl-proline in diabetes-related kidney fibrosis. Sci. Rep. 2016, 6, 29884. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; Grahovac, G.; Zeisberg, M.; Kalluri, R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 2007, 56, 1825–1833. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Li, J.; Kitada, M.; Fujita, H.; Yamada, Y.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Sirt3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 2018, 9, 997. [Google Scholar] [CrossRef]
- Zhou, D.; Li, B.H.; Wang, J.; Ding, Y.N.; Dong, Y.; Chen, Y.W.; Fan, J.G. Prolyl oligopeptidase inhibition attenuates steatosis in the l02 human liver cell line. PLoS ONE 2016, 11, e0165224. [Google Scholar] [CrossRef] [Green Version]
- Scheen, A.J. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab. 2004, 30, 487–496. [Google Scholar]
- Jacobsen, P.; Andersen, S.; Jensen, B.R.; Parving, H.H. Additive effect of ace inhibition and angiotensin ii receptor blockade in type i diabetic patients with diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 992–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauer, M.; Zinman, B.; Gardiner, R.; Suissa, S.; Sinaiko, A.; Strand, T.; Drummond, K.; Donnelly, S.; Goodyer, P.; Gubler, M.C.; et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N. Eng. J. Med. 2009, 361, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Neldam, S.; Tikkanen, I.; Oren, S.; Viskoper, R.; Watts, R.W.; Cooper, M.E. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: The candesartan and lisinopril microalbuminuria (calm) study. Bmj 2000, 321, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.; Schmieder, R.E.; McQueen, M.; Dyal, L.; Schumacher, H.; Pogue, J.; Wang, X.; Maggioni, A.; Budaj, A.; Chaithiraphan, S.; et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ontarget study): A multicentre, randomised, double-blind, controlled trial. Lancet 2008, 372, 547–553. [Google Scholar] [CrossRef]
- Kunz, R.; Friedrich, C.; Wolbers, M.; Mann, J.F. Meta-analysis: Effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann. Intern. Med. 2008, 148, 30–48. [Google Scholar] [CrossRef]
- Mauer, M.; Fioretto, P. Preventing microalbuminuria in type 2 diabetes. N. Eng. J. Med. 2005, 352, 833–834, author reply 833–834. [Google Scholar]
- Wu, H.Y.; Peng, C.L.; Chen, P.C.; Chiang, C.K.; Chang, C.J.; Huang, J.W.; Peng, Y.S.; Tu, Y.K.; Chu, T.S.; Hung, K.Y.; et al. Comparative effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin ii receptor blockers for major renal outcomes in patients with diabetes: A 15-year cohort study. PLoS ONE 2017, 12, e0177654. [Google Scholar]
- Mavridis, D.; Palmer, S.C.; Strippoli, G.F. Comparative superiority of ace inhibitors over angiotensin receptor blockers for people with ckd: Does it matter? Am. J. Kidney Dis. 2016, 67, 713–715. [Google Scholar] [CrossRef]
- Baltatzi, M.; Savopoulos, C.; Hatzitolios, A. Role of angiotensin converting enzyme inhibitors and angiotensin receptor blockers in hypertension of chronic kidney disease and renoprotection. Study results. Hippokratia 2011, 15, 27–32. [Google Scholar]
- Wu, H.Y.; Huang, J.W.; Lin, H.J.; Liao, W.C.; Peng, Y.S.; Hung, K.Y.; Wu, K.D.; Tu, Y.K.; Chien, K.L. Comparative effectiveness of renin-angiotensin system blockers and other antihypertensive drugs in patients with diabetes: Systematic review and bayesian network meta-analysis. BMJ 2013, 347, f6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Ritz, E. Combination therapy with ace inhibitors and angiotensin ii receptor blockers to halt progression of chronic renal disease: Pathophysiology and indications. Kidney Int. 2005, 67, 799–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, T.; Ma, L.J.; Yang, H.; Zuo, Y.; Tang, Y.; Han, J.Y.; Kon, V.; Fogo, A.B. Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. Am. J. Physiol. Ren. Physiol. 2010, 298, F683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matavelli, L.C.; Huang, J.; Siragy, H.M. Angiotensin at(2) receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 2011, 57, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 2000, 35, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Danyel, L.A.; Schmerler, P.; Paulis, L.; Unger, T.; Steckelings, U.M. Impact of at2-receptor stimulation on vascular biology, kidney function, and blood pressure. Integr. Blood Press Control 2013, 6, 153–161. [Google Scholar]
- Padia, S.H.; Carey, R.M. At2 receptors: Beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch. 2013, 465, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shi, S.; Srivastava, S.P.; Kitada, M.; Nagai, T.; Nitta, K.; Kohno, M.; Kanasaki, K.; Koya, D. Fgfr1 is critical for the anti-endothelial mesenchymal transition effect of n-acetyl-seryl-aspartyl-lysyl-proline via induction of the map4k4 pathway. Cell Death Dis. 2017, 8, e2965. [Google Scholar] [CrossRef]
- Wang, B.; Jha, J.C.; Hagiwara, S.; McClelland, A.D.; Jandeleit-Dahm, K.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2013. [Google Scholar]
- Macconi, D.; Tomasoni, S.; Romagnani, P.; Trionfini, P.; Sangalli, F.; Mazzinghi, B.; Rizzo, P.; Lazzeri, E.; Abbate, M.; Remuzzi, G.; et al. Microrna-324-3p promotes renal fibrosis and is a target of ace inhibition. J. Am. Soc. Nephrol. 2012, 23, 1496–1505. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Shi, S.; Koya, D.; Kanasaki, K. Lipid mediators in diabetic nephropathy. Fibrog. Tissue Rep. 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.P.; Koya, D.; Kanasaki, K. Micrornas in kidney fibrosis and diabetic nephropathy: Roles on emt and endmt. Biomed. Res. Int. 2013, 2013, 125469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, H.; Liu, J.; Han, P.; Li, X.; Bai, H.; Zhang, C.; Sun, X.; Teng, Y.; Zhang, Y.; et al. Variations in microrna-25 expression influence the severity of diabetic kidney disease. J. Am. Soc. Nephrol. 2017, 28, 3627–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Wan, J.; Hou, X.; Geng, J.; Li, X.; Bai, X. Microrna-27a promotes podocyte injury via ppargamma-mediated beta-catenin activation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Khan, S.Q.; Khaliqdina, S.; Altintas, M.M.; Grahammer, F.; Zhao, J.L.; Koh, K.H.; Tardi, N.J.; Faridi, M.H.; Geraghty, T.; et al. Absence of mir-146a in podocytes increases risk of diabetic glomerulopathy via up-regulation of erbb4 and notch-1. J. Biol. Chem. 2017, 292, 732–747. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Koh, P.; Winbanks, C.; Coughlan, M.T.; McClelland, A.; Watson, A.; Jandeleit-Dahm, K.; Burns, W.C.; Thomas, M.C.; Cooper, M.E.; et al. Mir-200a prevents renal fibrogenesis through repression of tgf-beta2 expression. Diabetes 2011, 60, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Zhong, X.; Huang, X.R.; Meng, X.M.; You, Y.; Chung, A.C.; Lan, H.Y. Microrna-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 2014, 22, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Krupa, A.; Jenkins, R.; Luo, D.D.; Lewis, A.; Phillips, A.; Fraser, D. Loss of microrna-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 438–447. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs. Genes 2020, 11, 211. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020211
Srivastava SP, Goodwin JE, Kanasaki K, Koya D. Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs. Genes. 2020; 11(2):211. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020211
Chicago/Turabian StyleSrivastava, Swayam Prakash, Julie E. Goodwin, Keizo Kanasaki, and Daisuke Koya. 2020. "Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs" Genes 11, no. 2: 211. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11020211