History of DNA Helicases

1
Section on DNA Helicases, Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA
2
Department of Biology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
*
Authors to whom correspondence should be addressed.
Genes 2020, 11(3), 255; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030255
Submission received: 27 January 2020
/
Revised: 18 February 2020
/
Accepted: 20 February 2020
/
Published: 27 February 2020
(This article belongs to the Special Issue DNA Helicases: Mechanisms, Biological Pathways, and Disease Relevance)
Abstract
:Since the discovery of the DNA double helix, there has been a fascination in understanding the molecular mechanisms and cellular processes that account for: (i) the transmission of genetic information from one generation to the next and (ii) the remarkable stability of the genome. Nucleic acid biologists have endeavored to unravel the mysteries of DNA not only to understand the processes of DNA replication, repair, recombination, and transcription but to also characterize the underlying basis of genetic diseases characterized by chromosomal instability. Perhaps unexpectedly at first, DNA helicases have arisen as a key class of enzymes to study in this latter capacity. From the first discovery of ATP-dependent DNA unwinding enzymes in the mid 1970’s to the burgeoning of helicase-dependent pathways found to be prevalent in all kingdoms of life, the story of scientific discovery in helicase research is rich and informative. Over four decades after their discovery, we take this opportunity to provide a history of DNA helicases. No doubt, many chapters are left to be written. Nonetheless, at this juncture we are privileged to share our perspective on the DNA helicase field – where it has been, its current state, and where it is headed.
1. Discovery of DNA Unwinding Enzymes and Coining the Term Helicase
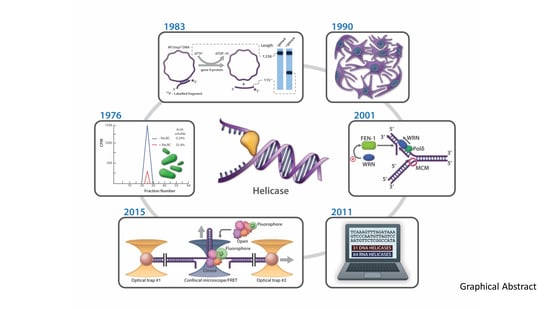
The discovery of proteins capable of ATP-dependent enzymatic unwinding of duplex DNA was first reported in 1976 by Hoffmann-Berling and colleagues at the University of Heidelberg [1,2] and Mackay and Linn at the University of California, Berkeley [3]. As pointed out recently in a review by Lohman and Fazio [4], the term “helicase”, referring to an ATP-dependent duplex DNA unwinding enzyme, was coined by Hoffmann-Berling in 1978 [5] and appeared in two subsequent publications in 1979 [6,7]. Even at this early stage, a suggestion was made that different helicase enzymes were unique in terms of properties and mechanism of action (e.g., processivity). Mackay and Linn described E. coli RecBC as an ATP-dependent unwinding enzyme in which catalytic duplex strand separation was detectable (under conditions where the nuclease is suppressed by a DNA binding protein), leading them to hypothesize that “the major contribution of RecBC enzyme to recombination would be the unwinding of DNA” [3]. Thus, from both biochemical and genetic viewpoints, the discovery of DNA helicases immediately provoked speculation regarding their mechanisms and biological roles. A timeline for the discovery of prokaryotic, eukaryotic, and viral DNA helicases is shown in Figure 1. Bacteriophage and plant DNA helicases were isolated not long after the discovery of the first bacterial helicases but it would be approximately 10 years until mammalian or yeast DNA helicases were reported. DNA helicase activities from mouse cell extracts were reported in several studies in the late 1980’s and early 1990’s [8,9,10]. The Falaschi laboratory at the International Centre for Genetic Engineering and Biotechnology (Trieste, Italy) isolated the first DNA helicase from human cells in 1990 [11]. Subsequently, DNA helicases were isolated from bovine mitochondria [12] and pea chloroplasts [13], indicating their ubiquitous presence. As of 2011, there were an estimated 95 helicases or putative helicases encoded by the human genome; 31 DNA helicases and 64 RNA helicases [14]. Molecular and cellular studies have elucidated the functional aspects of numerous DNA helicases in various pathways of nucleic acid metabolism. Fifteen years ago, Narendra and Renu Tuteja provided a historical account of prokaryotic and eukaryotic DNA helicases in an informative review [15]. Here, we will highlight some of the major advances in understanding helicase mechanism and function over the years as marked by continued progress in the field.
2. Helicase Superfamilies Defined by Sequence Homology and Structure
In 1982, Walker et al. of The MRC Centre (Cambridge, UK) described, from their sequence analysis of α- and β-subunits of ATP synthase, two clustered amino acid regions which shared strong similarity with other ATP-binding proteins [25]. These proteins included a known bacterial ATPase, the recombination protein RecA, adenylate kinase, and myosin from both nematode and rabbit. This led them to propose a common nucleoside triphosphate (NTP) binding fold present in each of these proteins. Six years later, in May of 1988, two reports appeared in Nature bringing helicases into the picture. In the first, Gorbalenya and colleagues described an alignment of the amino acid sequence corresponding to the common nucleotide binding fold (proposed by Walker et al., 1982) present in the bacterial RecD protein (a helicase subunit of the RecBCD DNA helicase complex) with a viral NTP motif-containing RNA helicase [26]. In the same issue of Nature, Hodgman commented on the presence of seven conserved amino acid motifs found in a superfamily (SF) of proteins implicated in DNA replication and recombination, including the E. coli DNA helicases UvrD, Rep, RecB, and RecD [27]. Shortly after, Lane added eukaryotic translation initiation factor eIF-4A, known to be a RNA helicase, and the putative helicase p68 to the classification [28], suggesting an even larger family of helicases or helicase-like proteins. Based on these early visual and computer-aided alignments, the helicase SF consisted of proteins which shared seven distinct highly conserved amino acid segments, or helicase motifs, comprising the two sites (designated A and B) for NTP-binding as they came to be commonly referred [29]. Further sequence alignment revealed a more refined classification of two related SFs with different sets of helicases or putative helicases [30]. To date, six SFs of helicases, putative helicases, or DNA translocases (which lack the classic NTP-driven strand separation activity typical of helicase) have been defined with discovery of new genes [31], and are nicely discussed in a review by Singleton et al. [32].
Site-directed mutagenesis has proven to be a powerful approach to interrogate the biochemical and genetic importance of conserved residues within the helicase motifs. One of the earliest such studies from the Prakash laboratory demonstrated that mutation of the invariant lysine within motif I (ATPase A site) of the S. cerevisiae Rad3 helicase abolished its ATPase and unwinding activities, and rendered it inactive in a key post-incision step of nucleotide excision repair (NER) necessary for the removal of ultraviolet (UV) light-induced pyrimidine dimers [33]. Similar mutations in the ATPase A site were found to catalytically inactivate the bacterial PriA [34] and UvrD [35] helicases as well; however, the biological consequences were distinct. Characterization of site-specific mutations in motifs other than the classic ATPase A and B sites have been informative. For example, experimental evidence from the Matson, Wigley, and Keck laboratories demonstrated that motif III serves an important function in coupling DNA binding with ATP binding/hydrolysis and strand separation that is necessary for the efficient catalytic cycles of UvrD [36], PcrA [37], and RecQ [38] helicases, respectively. For further reading about helicase motif structure-function studies and structural perspectives, see [39,40,41].
3. Biochemical Studies of DNA Helicases and Their Substrate Specificity
Over the years, remarkable progress has been made in developing experimental techniques to detect and measure helicase-catalyzed DNA unwinding. Figure 2 shows representative helicase assays illustrating different biochemical and biophysical strategies that have been utilized over time. Of course, there are many applications and experimental techniques for studying DNA helicases which we are unable to address due to space limitations and the enormity of the field which continues to grow. In the immediate sections we will describe some fundamental lessons learned about the properties and mechanisms of DNA helicases using novel experimental procedures and strategies. A special section found later in the text highlights single-molecule (SM) approaches that are gaining in popularity to measure helicase activity.
3.1. Helicase Directionality
Early studies of DNA helicase enzymes focused on their single-stranded DNA (ssDNA) stimulated ATPase activity in addition to their helicase activity [1,2,6,7]. This, together with the apparent requirement of ssDNA for a helicase to catalyze duplex DNA unwinding, suggested an interaction with ssDNA leading to the notion of directionality of DNA translocation and unwinding. By its very nature, the DNA double helix is defined by two DNA strands with opposite chemical polarities wrapped around each other in a helical fashion and interacting through a series of noncovalent hydrogen bonds between the two strands [16]. The polarities of the two DNA strands are determined by the sugar-phosphate backbone in which the oxygen atom of the phosphodiester bond is covalently bonded to either the 3’ or 5’ positions of the intervening ribose moiety. The two strands of the DNA double helix run in opposite directions, dictating its anti-parallel nature. This structural feature of the DNA double helix has profound implications for its metabolism, including the copying of the genetic information by the process of semi-conservative DNA replication.
Unidirectional translocation by a helicase along ssDNA, fueled by the energy of nucleoside triphosphate hydrolysis, would in principle provide a mechanism whereby the directionality of duplex DNA unwinding would be established. Indeed, several laboratories established the directionality for the bacteriophage helicases T7 gene 4 protein and T4 gene 41 protein, as well as the bacterial Rep helicase (reviewed in [47]). In a seminal study by Yarranton and Gefter in 1979 radiolabeled partial duplex DNA substrates with defined polarities were incubated with purified Rep protein and ATP followed by ssDNA-specific S1 nuclease digestion (see Figure 2A) and DNA precipitation by trichloroacetic acid. Acid insoluble radioactivity was quantified to determine the fraction of duplex DNA susceptible to nuclease and to infer if a partial duplex substrate with a 3’ or 5’ single-stranded tail was unwound. These experiments indicated that Rep translocates 3’ to 5’ along the single-stranded region of the substrate to reach and unwind the adjacent duplex [48].
Although assays which relied on nuclease sensitivity or isolation of products by sucrose gradient or bound to nitrocellulose were useful tests for helicase-catalyzed DNA unwinding [1], the choice of DNA substrates was narrow and experimental techniques were tedious and relied on indirect measurements. For improved quantitation and direct visualization of helicase reaction products, a more straightforward assay was in demand. The first direct demonstration that a DNA helicase employs its unidirectional translocation on ssDNA to unwind duplex DNA in a directionally specific manner was provided by Nancy Nossal’s laboratory at the NIH in 1982 [20]. A radiolabeled partial duplex DNA substrate characterized by two different length duplex DNA fragments separated by an intervening long ssDNA tract was incubated with the T4 gene 41 protein in the presence of ATP and the reaction products were resolved on a non-denaturing polyacrylamide gel. Using this technique, it was demonstrated that T4 gene 41 moves in a 5’ to 3’ direction with respect to the strand on which it is bound. In 1983 Matson et al. described a similar DNA substrate in which a complementary strand of a radioactively labeled denatured DNA fragment of defined size was annealed to circular single-stranded M13 viral DNA (Figure 2B). Here the substrate had an additional feature in that it possessed a noncomplementary 3’-tail, thereby resembling a forked duplex [42]. This DNA substrate, and its derivatives in which the length of the noncomplementary single-stranded tail was varied, enabled the authors to determine the minimal DNA substrate requirements of T7 gene 4 protein to unwind the DNA duplex adjacent to the junction. The advantage of a direct helicase assay to measure DNA unwinding was convincingly evidenced by the demonstration that the bacterial UvrD helicase, an enzyme with a vital role in both NER and mismatch repair (MMR), translocates 3’ to 5’ in its unwinding mechanism [49], opposite to the directionality of T4 gene 41 or T7 gene 4 helicase proteins. As discussed by Matson in that paper, the 3’ to 5’ directionality of UvrD helicase has implications for the mechanisms of both the MMR and NER pathways.
The mechanism(s) whereby a given DNA helicase achieves unidirectional polarity of movement on DNA gained considerable interest in the late 1990’s. Some insight into the mechanism of helicase DNA translocation and directionality was garnered from structural and biochemical studies. Relative DNA binding affinity of the two RecA motor domains and protein contacts with ssDNA of a defined polarity are likely to dictate directionality of helicase movement coupled to ATP hydrolysis, as suggested by structural studies of the B. stearothermophilus PcrA helicase [50,51] and the E. coli Rep helicase [52]. The precise mechanistic details are likely to be different depending on the helicase.
In 2009, the Wigley lab published a paper that provided a mechanistic basis for how the sequence-related D. radiodurans DNA helicase RecD2 translocates on DNA with an opposite polarity (5’ to 3’) to that of PcrA. Their studies suggested that relative grip strength of defined protein domains in the helicase dictate its directionality of movement as opposed to opposite orientations of ssDNA binding sites [53]. This theme resurfaced in research on the XPD helicase from the Kisker and Spies laboratories who published papers in 2012 which shed light on its 5’ to 3’ directionality of translocation. The co-crystal structure of T. acidophilum XPD with a single-stranded oligonucleotide showed the trail of the translocated strand as it traversed the helicase pore; a comparison of the XPD structure with structures of DNA helicases that translocate with the opposite polarity (3’ to 5’) indicated the same orientation of DNA binding by the two RecA domains, suggesting that a reversal of protein gripping is responsible for the different polarities [54]. Studies that mapped XPD protein contacts on DNA demonstrated that T. acidophilum XPD binds ssDNA with an orientation similar to DNA helicases that translocate 3’ to 5’; one of the RecA domains in collaboration with the iron-sulfur (Fe-S) domain most likely provides the wedge for duplex separation [55].
3.2. Helicases Resolve Unconventional DNA Structures
In early experiments helicase-catalyzed DNA unwinding was assessed using partial duplex DNA substrates with a single-strand tail adjacent to the duplex region to allow helicase loading. However, these simplistic double-stranded DNA (dsDNA) molecules do not reflect the in vivo situation. An example, noted above, is the T7 gene 4 protein which requires a forked duplex DNA molecule [42]. Moreover, nucleic acid metabolic processes that occur in cells are relevant. During transcription stable DNA:RNA hybrids may form, causing one of the strands of the original DNA duplex to be displaced. These so-called R-loops may potentially interfere with the progress of an advancing replisome, suggesting that their timely resolution is important for genomic stability [56]. A role for a DNA helicase in this capacity was first suggested by experimental evidence that UvrD unwinds stable DNA:RNA hybrids even more efficiently than DNA duplexes in vitro [57]. The poor ability of RNA to serve as an effector for UvrD-catalyzed ATP hydrolysis [58] suggests a DNA-specific loading and interaction mechanism for unwinding the DNA:RNA hybrid. Supporting this general principle, to our knowledge the vast majority of DNA helicases characterized to date which bind DNA with higher affinity than RNA also preferentially unwind DNA duplex substrates compared to RNA duplex substrates. In addition to UvrD, the replicative bacterial, archaeal, and eukaryotic DNA helicases can unwind RNA-DNA substrates [59].
Over time, researchers investigated the activity of select helicases on nucleic acid structures that arise in specific pathways of cellular DNA metabolism. One such substrate is the Holliday Junction (HJ), a branched DNA molecule with four double-stranded arms that represents a key intermediate of genetic recombination and double-strand break repair that occurs during the S/G2 phases of the cell cycle. Building on genetic studies from Bob Lloyd’s laboratory demonstrating the importance of E. coli RuvA, RuvB, and RuvC in recombinational repair [60] and the finding that RuvC interacts with HJs to catalyze their symmetrical endonucleolytic cleavage [61], Steve West’s laboratory determined that the ATPase/helicase RuvB together with the DNA binding protein RuvA catalyzed HJ branch-migration in an ATP-dependent manner [62]. Thus, the RuvA/B helicase complex was determined to be capable of interacting with and branch-migrating a HJ structure that lacked any extensive ssDNA character. This discovery was significant and set a precedent for later work showing that human RecQ helicases involved in homologous recombination (HR) repair and implicated in diseases of premature aging and cancer (e.g., BLM [63], WRN [64]) also bind tightly to and branch-migrate HJs in an ATP-dependent manner [65] (see Genetic Diseases, Section 10). It should be noted that UvrD helicase also binds and unwinds HJ DNA substrates [66] likely explaining (at least partially) the recombination phenotype associated with uvrD mutants.
Although it is generally true that a given DNA helicase requires a ssDNA loading tail to catalyze unwinding of the adjacent duplex DNA (at least in vitro), there are exceptions. At relatively high helicase protein concentrations, E. coli RecQ was reported to unwind blunt duplex DNA substrates [67]. Thermal breathing or fraying of the duplex end promoted by helicase binding to the DNA substrate may play a role in this reaction. UvrD helicase, at elevated protein concentrations, was also found to initiate unwinding of duplex DNA from blunt ends as well as nicks [68]. The latter may provide an entry site for UvrD during DNA MMR [69,70] or NER [69,71]. Although a role for UvrD in post-incision strand release in bacterial MMR was elucidated in the early 1990s, we still do not know the identity of the helicase(s) (if any) implicated in this strand release step of eukaryotic MMR.
In other work, it was observed that the human RecQ 3’ to 5’ DNA helicases WRN [72], BLM [73,74], and RECQL1 [75] efficiently unwind 5’ flap DNA substrates despite the absence of a pre-existing 3’-ssDNA tail (Figure 3A). These studies, supported by mechanistic experiments with WRN helicase [72], suggest that junction-specific DNA binding and strand-specific loading are important properties of certain DNA helicases. The 5’ flap substrate is an important intermediate during Okazaki fragment maturation or strand displacement DNA synthesis in base excision repair. The coordinate action of human RecQ helicases with structure-specific Rad2 nucleases (FEN-1, EXO1) on 5’ flap DNA substrates is detailed in a number of studies (reviewed in [76]) (Figure 3A). Helicases also act upon unusual DNA structures that deviate from the canonical DNA double helix. DNA triplexes, G-quadruplexes (G4), and left-handed Z-DNA are prone to form at specific DNA sequences [77]. Such alternatively arranged DNA conformations potentially influence replication and transcription. In addition, these alternate DNA structures are proposed to form at specialized regions of chromosomal DNA (e.g., G4: guanine-rich telomeres, promoter elements, ribosomal DNA; triplex DNA: polypurine/polypyrimdine tracts; Z-DNA: alternating G/C sequence elements) and have consequences for genomic stability. Over the years, a number of groups have obtained biochemical evidence that DNA helicases resolve triplex DNA and G-quadruplex DNA in vitro (see Table 1 for representative examples).
Although beyond the scope of this review, biological and molecular evidence suggest that certain DNA helicases preserve genomic stability and maintain cellular homeostasis by resolving such alternate DNA structures. G4-resolving helicases are discussed in recent reviews [78,79]. A seminal study from the Lansdorp laboratory first suggested that a putative DNA helicase known as DOG-1 in C. elegans suppressed the accumulation of G4 structures in the lagging strand ssDNA template thereby preventing deletions in regions of the genome characterized by guanine-rich DNA [80]. Later studies substantiated important and conserved roles of DOG-1 homologs (FANCJ) in G4 DNA metabolism [81,82,83] (Figure 3B), including epigenetic regulation [84,85]. For an interesting perspective on FANCJ and the G4-resolving helicases RTEL1 and BLM, readers are encouraged to read reference [86].
Evidence suggests that Pif1 family helicases from species ranging from bacteria to human [87] resolve G4 DNA structures in vitro [88,89,90,91,92,93,94,95]. A role of PIF1 in G4 DNA metabolism in vivo is perhaps best exemplified by S. cerevisiae Pif1 [89,90,91,96], the founding member of the Pif1 family of helicases. Recent results from a live-cell imaging approach demonstrated that replication rates are significantly reduced by genomic DNA sequences predicted to form G4 in the lagging strand template but not the leading strand template in individual yeast cells [97]. The stimulatory effect of Pif1 on DNA replication was dependent on its interaction with PCNA, suggesting that Pif1 G4 resolution coupled with progression of the replication machinery is crucial for efficient DNA synthesis of the lagging strand (Figure 3C).
As elaborated upon below in Section 9.2, experimental evidence suggests that the XPB and XPD helicases are involved in transcriptional regulation by their interactions with G4 DNA structures associated with gene promoters [98].
4. Proposed Models for Helicase-Catalyzed DNA Unwinding
As it became apparent in the late 1970s and early 1980s that there are numerous DNA helicases with defined biochemical properties and DNA substrate specificities, an immense interest in characterizing their mechanisms of action developed. This led to more refined studies of helicase proteins, incorporating biochemical and biophysical approaches. Not only were researchers investigating protein assembly states but also changes in the conformation and functionality of the helicase that were induced by NTP and DNA binding. Efforts in these areas, along with new information on the structures of DNA helicase proteins, fueled even greater attention to mechanisms for DNA unwinding. In the following sections, we will discuss some prominent models for helicase-catalyzed DNA unwinding.
4.1. Rolling Model for Rep Helicase-Catalyzed Duplex DNA Unwinding
Significant advances in mechanistic studies of DNA helicases kicked into high gear in the late 1980s and early 1990s with the availability of abundant quantities of highly purified homogenous recombinant helicase protein that had been overexpressed in bacteria [125]. Research from Tim Lohman’s laboratory initially focused on the molecular mechanism of E. coli Rep helicase. Using biophysical and biochemical techniques, the Rep assembly state and the effects of nucleotide binding on Rep’s interaction with ssDNA or dsDNA were examined. Based on their studies, Wong and Lohman proposed a rolling model for Rep-catalyzed duplex DNA unwinding in which allosteric effects of nucleotide binding influenced the protein’s interaction with ssDNA or duplex DNA thereby providing a means for Rep to be continuously bound to the DNA substrate it was actively unwinding [126]. The data leading to the proposal of the rolling model is also consistent with the inch-worm model (see below), commonly accepted today. This advance for understanding helicase mechanism suggested that conformational changes (induced by nucleotide and DNA binding [127]) would affect oligomerization state, unwinding activity, and helicase processivity. A series of papers detailing kinetic analyses of Rep helicase suggested the functional cooperativity of Rep monomers, supporting a dimeric mode of action for Rep helicase that may be applicable to other DNA helicases (for review, see [128]). For example, the dimeric form of the FANCJ helicase (implicated in Fanconi Anemia (FA) see Genetic Diseases section, Section 10) was found to have a greater specific activity for ATPase, DNA binding, and helicase, compared to the monomer [129]. Further studies of FANCJ oligomerization should address the role of ligand-induced conformational changes in its mechanism of unwinding. Moreover, a structural determination of FANCJ, as well as the sequence-related DNA helicases RTEL1 and DDX11, and their interactions with ssDNA and dsDNA will be helpful.
4.2. Inchworm Model for PcrA Helicase-Catalyzed Duplex DNA Unwinding
In contrast to the evidence for Rep dimerization, the sequence-related PcrA helicase from B. stearothermophilus was determined by Bird et al. at the University of Oxford to exist solely as a monomer in 1998 [130]. This observation led the authors to propose that PcrA’s mechanism of DNA unwinding was distinct from that of a rolling model involving two (or more) subunits. Harkening back to an earlier related model of helicase action proposed by Yarranton and Gefter [48], the ability of PcrA to interact simultaneously with both ssDNA and dsDNA and additional evidence prompted Dale Wigley’s laboratory to propose an inchworm model for PcrA unwinding in which the monomeric helicase extends into duplex DNA as it translocates in an ATP-dependent fashion [130]. The step size for duplex unwinding is inferred to be as short as 1 base pair (bp) for an inchworm helicase, whereas a helicase that operates by a rolling model may have a longer step size reflecting the DNA binding site size for each of the monomers.
Subsequent structural work from the Wigley laboratory demonstrated that the PcrA helicase monomer is capable of binding ssDNA and duplex DNA simultaneously when ATP, modeled by the ATP analog ADPNP, is bound to the enzyme [51]. In this mode, PcrA assumes a conformation in which the relative positions of the two RecA domains favor close interaction of the protein surface to duplex DNA. When ATP is hydrolyzed, modeled by a bound sulfate ion, the two RecA domains of PcrA assume a rigid conformation that reflects a conformation of the helicase protein complexed solely with ssDNA. Thus, a conformational change in PcrA induced by intrinsic ATP hydrolysis causes the helicase protein to dissociate from duplex DNA but remain bound to ssDNA. Consecutive cycles of DNA translocation and destabilization of duplex patches as short as 1 bp controlled by conformational changes in PcrA elicited by nucleotide binding and hydrolysis constitute the basic actions of the inchworm mechanism.
Interestingly, seven years later the Yang laboratory proposed a combined wrench-and-inchworm mechanism for unwinding by the sequence-related UvrD helicase in which binding of a single ATP molecule results in 1 bp of duplex DNA unwound and the subsequent release of ADP and Pi allows translocation of UvrD on the emerging single-strand [131].
4.3. Helicase Processivity, Functional Cooperativity, and Protein Displacement
Although the assembly state of helicases has been of considerable interest over the years, functional cooperativity of multiple helicase molecules in unwinding a given duplex DNA substrate may involve elements other than protein oligomerization. For example, UvrD helicase unwinds partial duplex DNA substrates of increasing duplex lengths by a protein concentration dependent mechanism in which the number of bp unwound is directly proportional to the helicase concentration in the reaction mixture [58]. Although not formally tested, a model was proposed suggesting that UvrD helicase molecules concentrate at the ssDNA: dsDNA junction, allowing unwinding of longer duplex lengths in a non-processive fashion. The limited helicase processivity of UvrD may be tailored for its functions in NER or MMR, which require the unwinding of relatively short duplex DNA tracts as compared with unwinding at the replication fork in which many thousands of bp are typically encountered by the bacterial replicative helicase DnaB (see Replicative Helicase section below). Not all helicases lack processivity. For example, the TraI helicase, which functions in conjugative DNA transfer, is a highly processive helicase that functions as a monomer to unwind duplex DNA tracts greater than 850 bp [132].
Kevin Raney’s laboratory has a long-standing interest in the requirements for optimal unwinding of duplex DNA molecules by helicase proteins. Using pre-steady state kinetic measurements, they determined that the hepatitis C virus helicase NS3 unwinds long DNA tracts by aligning multiple NS3 molecules on the ssDNA on which it translocates, visualized conceptually by an array of locomotives advancing together on a train track [133]. In contrast to this model for functional cooperativity among NS3 helicase molecules, experimental evidence suggests that trailing Dda helicase molecules (from phage T4) do not exert a significant effect on the leading Dda monomer for unwinding conventional duplex DNA substrates [134]. Therefore, unique differences in the train helicase model are likely to exist.
Roadblocks to helicase translocation, such as protein bound to the DNA, may evoke unique mechanisms of helicase action as it was reported that trailing Dda helicase molecules contacting the lead Dda molecule serve to facilitate protein displacement [135]. This functional cooperativity among Dda helicase molecules to displace protein bound to DNA is a mechanism that potentially contributes to the ability of Dda to dissociate a stationary RNA polymerase and allow a faster rate of fork movement by the T4 bacteriophage replication machinery, as originally shown in a landmark 1983 paper by Bedinger et al. [136]. The demonstration that Lac repressor protein bound tightly to the double-stranded region of a partial duplex DNA substrate exerts differential effects on the ability of various DNA helicases to displace it and unwind the underlying duplex suggests that certain helicases may have specialized roles in clearing protein roadblocks to DNA replication [137]. The McGlynn laboratory has made considerable progress in this area, providing evidence that accessory bacterial DNA helicases (e.g., Rep, UvrD) can enable the replicative DnaB helicase to bypass protein obstacles assuring smooth replisome progression [138]. For a review on this topic, see [139]. Biochemical studies demonstrate that the eukaryotic ssDNA binding protein RPA, which physically interacts with several SF2 RecQ and Fe-S helicases and increases their duplex DNA unwinding activity [140,141,142,143], stimulates helicase-catalyzed protein displacement [144]. Certain DNA helicases (e.g., yeast Srs2) displace the major strand exchange protein Rad51 bound to DNA [145,146], a function that is important to manage or control HR.
4.4. Strand Exclusion Model for Replicative Helicases with Multimeric Ring-Like Structures
The prototypical replicative helicases are represented by the bacterial DnaB and eukaryotic minichromosome maintenance (MCM) helicases, which each form a hexameric ring-like structure of the same (DnaB) or different (Mcm2–7) subunits (for review, see [147]). Both DnaB and MCM helicases are believed to unwind dsDNA by inserting one strand of the unwound duplex inside the donut ring and the other outside the ring. This mechanism is classically known as steric exclusion. However, recent studies suggest that ring-like helicases are characterized by subtle unique properties in terms of their interactions with the “excluded” strand [148]. Interestingly, the bacterial DnaB helicase operates with a 5’ to 3’ directionality placing it on the lagging strand template whereas eukaryotic MCM translocates 3’ to 5’ on the leading strand template. It should be noted that replicative helicases can translocate dsDNA [59,149].
The discovery of DnaB helicase as an essential factor in E. coli chromosomal DNA replication was achieved through genetic analyses from several groups [150,151,152,153,154] before the helicase activity of the protein was demonstrated. These studies, and additional findings from cellular and biochemical analyses of bacteriophage replication, set the stage for two biochemists from The Johns Hopkins University to hypothesize that DnaB was responsible for processive unwinding of duplex DNA at the replication fork. LeBowitz and McMacken biochemically tested purified DnaB for ATP-dependent DNA unwinding activity on a M13 partial duplex DNA substrate with over 1000 bp of duplex DNA and two preformed forks [155]. They subsequently proposed a mechanistic model for the functions of the major replication proteins at an E. coli replication fork. Following that, a series of papers from Arthur Kornberg’s laboratory (which began with the finding that in a reconstituted system DnaB helicase function is required for initiation of DNA synthesis at the chromosomal origin [156]) and other laboratories provided a more complete model that served as a gold standard for future studies on the mechanism of eukaryotic DNA replication. For Kornberg’s perspective on the lessons learned during the pursuit of characterizing the mechanism of DNA replication and the enzymes involved, see [157].
As discussed in a comprehensive review by Bochman and Schwacha [158], the MCM genes were first discovered in S. cerevisiae by the Tye laboratory employing a screen to detect mutants that were defective in the regulation of replication initiation [159]. Subsequent studies in yeast [160,161,162] and X. laevis [163,164,165] provided further evidence for their involvement in various stages of cellular DNA replication. Much of the early work on the biochemical characterization of MCM ATPase/helicase function, assembly state, and interaction with nucleic acid was performed with the homologous archaeal proteins [166], which proved to be (and still are) highly informative for the analysis of eukaryotic MCM (see [158] for details). The first demonstration that the MCM protein complex has ATPase and DNA helicase activity came from studies by Yukio Ishimi [167]. The intricate mechanistic functions of the MCM helicase complex continue to be a fascinating area of detailed study offering new insights into its roles in DNA replication licensing, initiation, progression, and termination (for review, see [168,169,170]). Of particular interest in recent years is the mechanism(s) utilized by the MCM helicase complex to handle DNA roadblocks and the functional consequences for obstacle bypass (reviewed in [171]). Recently, a crystal structure of S. solfataricus MCM bound to ssDNA, an ATP analog, and ADP was solved which indicated a rotary mechanism to translocate DNA that is conserved for both archaeal and eukaryotic MCM helicases [172]. A model was suggested for the conversion of the MCM double-hexamer encircling dsDNA to single hexamers encircling ssDNA that bears relevance to replication initiation.
5. DNA Helicase Protein Structures
Structural information derived from biophysical approaches has provided important insights to mechanistic features of how various DNA helicases unwind duplex DNA. A summary of the major discoveries of prominent DNA helicase structures is shown in Table 2. This summary is not meant to be exhaustive but rather representative for members of the various helicase families and to provide the reader an opportunity to consult the cited references for more in-depth discussion of the findings. The first structural information for a DNA helicase that can be found in the literature is from the Hurwitz laboratory in 1989 in which they reported that the Simian virus 40 (SV40) large tumor (T) antigen, required for initiation of viral replication, forms ATP-dependent double hexamers around dsDNA as revealed by scanning transmission electron microscopy [173]. In 1995, a significant advance was made by the Egelman and Patel laboratories in their analysis of the bacteriophage T7 helicase/primase [174]. They utilized electron microscopy, three-dimensional reconstruction, and protein crosslinking to show that the T7 gene 4 protein forms a hexameric ring that encircles ssDNA. Moreover, they showed the protein is bound to the DNA with a defined polarity, suggesting a structural basis for the 5’ to 3’ directionality of helicase movement on DNA and the mode of unwinding. These and other studies provided the foundation for the field’s current appreciation that replicative helicases, including E. coli DnaB [175] and MCM [176,177], form ring-like structures around DNA to enact DNA unwinding. However, the molecular mechanics and functions of DNA unwinding by the replicative helicases (e.g., MCM) is informed by not only structural characterization but also SM and biological studies (for review, see [178]).
In 1996, Dale Wigley’s laboratory reported the first crystal structure of a DNA helicase, which revealed that PcrA helicase has two RecA-like domains that form a cleft for ATP binding [50]. This discovery was closely followed by the determination of the first structure of a DNA helicase (Rep) bound to DNA, which suggested major domain swiveling coupled to DNA translocation [52]. A subsequent study of PcrA bound to DNA in the presence of a nucleotide analog led Wigley’s team to further define the inchworm mechanism for helicase-catalyzed DNA unwinding by SF 1 helicases [51] that remains prevalent today (see Section 4.2). With advances in understanding the molecular architecture of SF1 helicases, the Keck laboratory at the University of Wisconsin Medical School solved the structure of a SF2 helicase, namely E. coli RecQ, in 2003 [179]. This work revealed the structures of two auxiliary domains (Zn2+ binding; winged helix) in addition to the conserved RecA-like domains implicated in nucleotide binding. The E. coli RecQ structure provided insight into the molecular defects caused by missense mutations in a human RecQ helicase linked to the genetic disease Bloom syndrome (BS), discussed below [180,181,182].
Mechanistic features of how the E. coli RecBCD helicase-nuclease processes double-strand breaks were elucidated by structural determination of the protein complex [189]. The bipolar helicase complex has the two motor subunits (RecB, RecD) on opposite strands at the fork of the duplex with the RecC subunit positioned to split the two strands. The architecture of the DNA:protein complex enables nucleolytic cutting by RecB in a manner that is orchestrated by RecC’s recognition of the recombinational hotspot (Chi site).
In 2008, three laboratories in different countries (Tainer in the United States; White in the United Kingdom and Kisker in Germany) independently reported the crystal structures of the NER helicase XPD from three different archaeal species, providing evidence for the structural importance of a conserved Fe-S cluster residing within the helicase core domain and acting as a wedge during duplex DNA unwinding [191,192,193]. Steady progress in understanding the structures of RecQ helicases has continued, highlighted by a 2016 paper in which experimental evidence provided by the Gileadi and Vindigni laboratories implicated a strand separation pin buttressed by a protein dimer interface in human RECQL1 to mediate duplex DNA separation [196].
Although the focus of structural studies has historically been to elucidate the molecular mechanism for unwinding dsDNA, in 2018 two papers appeared which addressed the structural elements and molecular details for resolving G-quadruplex DNA by the S. cerevisiae Pif1 [201] and C. sakazakii RecQ [202] helicases. This area of research is likely to blossom with the growing interest in the roles of unconventionally structured DNA in genome biology.
6. Single-Molecule Studies of Helicase-Catalyzed DNA Unwinding
Bulk biochemical measurements of DNA helicase activity have been highly informative for characterizing substrate specificity, modulation of catalytic activity by protein interactions and post-translational modifications, and structure-function studies of site-directed mutant helicase proteins. However, detailed mechanistic studies of helicase function required more sophisticated and sensitive assays. This led researchers to develop experimental strategies to perform SM studies in which the molecular interactions of a helicase protein with its DNA substrate could be determined from a single event as opposed to an average of many events. The first published studies of a DNA helicase using SM strategies were performed with the E. coli RecBCD helicase/nuclease. In 2001, two papers appeared in Nature describing SM experiments to assess RecBCD’s DNA translocation and unwinding functions [204,205]. The strategies employed in the two studies were different. Bianco et al. visualized single RecBCD molecules as they moved on dsDNA by optically trapping individual fluorescently labeled dsDNA molecules attached to polystyrene beads and monitoring fluorescent dye displacement from the DNA substrate by RecBCD as the DNA substrate was unwound [204]. Their results indicated that RecBCD is highly processive, unwinding duplex lengths of over 40,000 bp at a rate of ~970 bp per second. In the companion paper, Dohoney and Gelles used tethered-particle light microscopy to detect DNA unwinding by RecBCD at a resolution of ~100 bp with a biotin-tagged RecD subunit bound to polystyrene beads [205]. They observed that most individual RecBCD molecules move at a constant rate as they traverse the duplex, which in the study was determined to be greater than 1 kb in length, in a manner that translocation and unwinding are tightly coupled.
As mentioned earlier, many DNA helicases (e.g., UvrD, Rep) lack the processivity exhibited by the RecBCD complex. To investigate the molecular mechanism of DNA unwinding catalyzed by the Rep helicase, Ha et al. used an assay based on fluorescent resonance energy transfer (FRET) with donor and acceptor moieties attached to opposite strands of a duplex DNA substrate, which in turn was bound to a polymer-coated surface [206]. Structural changes to the DNA substrate could be assessed by a specialized microscope, enabling detection of DNA unwinding, helicase stalling, duplex rewinding, and re-initiation of DNA unwinding. This high-powered approach, and other innovative strategies in SM experimentation, ushered in a new era for mechanistic studies of DNA helicases. Some of the important and diverse innovations in SM studies of DNA helicases are discussed below from a historical perspective, but due to the broad scope of the field readers are encouraged to consult some timely reviews [207,208,209].
In 2004 the Croquette laboratory described an experimental strategy to monitor UvrD helicase activity at the SM level using a nicked duplex DNA molecule anchored on one end to a magnetic lead and the other to a glass surface [44] (Figure 2D). With a force applied to stretch the ssDNA molecule, they were able to measure extensions in the DNA attributed to duplex unwinding. Based on their experimental results, the authors suggested a model in which strand switching by UvrD during dsDNA unwinding elongation allows rapid reannealing behind the translocating helicase. Over the years, models that include unwound DNA strands rezipping have become popular, and evidence for this has been derived from varied SM strategies. For example, in 2005 Myong et al. published a paper describing a site-specific modification of the Rep protein which labeled it with a dye (donor) so that helicase movement could be measured by FRET as it translocated away from a site-specific DNA acceptor moiety [210]. Using this approach, they were able to show that Rep monomer undergoes repetitive shuttling on DNA induced by a duplex or protein bound to the DNA. Several nice reviews from prominent laboratories appeared at this time which summarized insights into helicase mechanisms garnered by SM studies [211,212,213].
A long-standing question in the field centered on whether the ring-like replicative helicases unwound duplex DNA using a passive or an active mechanism. The passive mechanism of unwinding relies on thermal fluctuations at the fork that make available ssDNA which is then trapped by the replicative helicase as it translocates along the DNA lattice. Alternatively, certain models for helicase-catalyzed duplex DNA unwinding suggested an active unwinding mechanism in which the helicase protein itself destabilized the duplex region at a ssDNA:dsDNA junction. Both mechanisms are dependent on NTP hydrolysis, but the salient features of unwinding are distinct. To address this, Johnson et al. performed SM studies using individual T7 gene 4 protein helicase hexamers with DNA substrates of defined nucleotide sequence subjected to a destabilizing force at the fork junction [214]. Their results were consistent with an active mechanism of DNA unwinding by T7 gene 4 helicase.
The efficient coupling of a replicative DNA polymerase with a helicase has been a topic of considerable interest over the years. Dating back to 1996, the Marians laboratory determined that a protein interaction between the E. coli DNA polymerase III holoenzyme and the DnaB replicative helicase is responsible for rapid replication fork movement [215]. In 2005, the Patel laboratory provided evidence for a mechanical coupling between the T7 DNA polymerase and helicase, demonstrating that DNA synthesis accelerates helicase-catalyzed DNA unwinding [216]. Consistent with this work, in 2007 Charles Richardson’s laboratory reported that T7 DNA polymerase and gene 4 helicase protein interact to form a highly processive replication fork complex [217].
Structural characterization of RecBCD by the Wigley lab [189], discussed above, helped to elucidate the mechanism of the helicase-nuclease complex. The two motor subunits (RecB, RecD) of the RecBCD complex move with opposite polarities posing the unique question of how their motors are coordinated and what happens when the helicase encounters a recombination hotspot DNA sequence known as Chi. SM analysis suggested a model of lead motor subunit switching to explain RecBCD’s biological roles [218]. In addition to playing a fundamental role in HR, RecBCD also degrades foreign DNA via its intrinsic nuclease activity.
Helicase unwinding rates may be slowed by proteins bound to DNA, as revealed by the Spies’ laboratory SM studies of the XPD helicase [219]. However, certain helicases, like PcrA, are well equipped to strip proteins (RecA filament) bound to DNA [220]. Remarkably, even large protein-DNA adducts cross-linked to the translocating strand can be bypassed by the large T antigen replicative helicase, as demonstrated by ensemble assays [221]. In 2013, Finkelstein and Greene contributed an insightful review detailing SM studies which helped to elucidate how DNA helicases deal with protein obstacles in their path [222]. Intricate details of how the CMG (Cdc45, Mcm2-7, GINS) helicase complex bypasses protein-DNA cross-links to allow their removal by proteolysis are emerging through combined experimental approaches that include SM [223].
Loading of the replicative Mcm2–7 helicase and initiation of DNA unwinding at origins of replication has attracted great interest in the field. Using a SM multi-wavelength fluorescence strategy with distinguishable fluorophores attached to different proteins and FRET-based measurements, Ticau et al. presented evidence in a landmark 2015 paper that bidirectional loading of the Mcm2-7 hexamer complexes is achieved by coordinated and distinct mechanisms to begin origin unwinding and DNA synthesis in opposing directions [224]. Loading of eukaryotic Mcm2-7 helicase complexes to origin-DNA occurs sequentially to form the double-hexamer, a parameter that was not definitively determined in the structural study of S. solfataricus MCM by Meagher et al. [172]. Ultimately, the combination of structural, biochemical and kinetic approaches with advanced techniques may provide further elucidation of the precise mechanism of action by MCM during origin unwinding and initiation of DNA synthesis.
Magnetic tweezers-based SM assays have allowed researchers to gain a better molecular understanding of DNA helicases (e.g., RecQ) that act upon specific DNA structures such as the strand invasion intermediate displacement (D)-loop that forms during HR [225]. Various steps in the HR pathway can now be studied at SM resolution with the development of a variety of imaging methods (for review, see [226]). However, measurement of helicase activity under applied force can introduce complexity to the unwinding mechanism that may deviate from physiological conditions. Therefore, studies like a very recent one published by the Galletto laboratory that investigated the DNA unwinding mechanism by Pif1 in real-time using SM Förster resonance energy transfer approaches will continue to be valuable [227].
Less than two decades of research by SM experimentation has provided a wealth of new insights to the molecular mechanisms of DNA helicases. This field has grown tremendously with more sophisticated techniques that have begun to interface with studies of complex DNA processes (e.g., DNA end resection, semi-conservative replication, genetic recombination, DNA repair, transcriptional activation). The modern applications of SM approaches, such as high-resolution “fleezers” [45,46] (Figure 2E), leave little doubt that tremendous advances in our understanding of how helicases mechanistically unwind complex DNA structures and collaborate with other nucleic acid metabolizing proteins are on the horizon.
7. Protein Interactions of DNA Helicases
In 1993 Aziz Sancar at the University of North Carolina at Chapel Hill described his molecular matchmaker model as it applied to both NER and MMR in E. coli [228]. This description helped pave the way for a whole field of study in protein interactions and their importance in DNA repair, and more broadly in nucleic acid transactions. Identification and characterization of physical and functional interactions between DNA helicases and other nuclear or mitochondrial proteins has accelerated an understanding of cellular and genetic pathways required for genomic stability, a robust DNA damage response, DNA replication, normal gene expression, and ultimately cellular homeostasis. The catalytic efficiency of DNA unwinding by helicases can be modulated by their physical and/or functional interactions with such factors as ssDNA binding proteins (e.g., E. coli SSB [229,230,231] or human RPA [140,141,232,233]), shelterin proteins that bind to telomeric DNA sequences (e.g., TRF2 [234]), and DNA polymerases that convert unwound ssDNA to duplex by nascent DNA synthesis [216], as well as many other proteins. Physical interaction of RPA with WRN or BLM helicases plays an important role in stimulation of helicase-catalyzed DNA unwinding [232]. Alternatively, helicases physically bind to nucleic acid metabolizing proteins and alter their functionality. For example, the cleavage efficiency of structure-specific Rad2 nucleases is greatly increased by physical interactions with certain RecQ DNA helicases [76]. T7 gene 4 stimulates DNA synthesis by T7 DNA polymerase [235], a functional interaction that has been found to be relevant for many helicase-DNA polymerase partnerships. The topic of helicase protein interactions with other factors is extensive and beyond the scope of this review, but the reader can find some very useful review articles in this area [236,237,238,239,240,241,242,243].
8. Post-Translational Modifications of Helicase Proteins
Experimental studies over the years have provided strong evidence that the functions of DNA helicases, their interactions with protein partners, and their protein stability can be affected by specific post-translational modifications. Modulation of helicase function by post-translational modification provides a cellular mechanism for acute responses. The FANCJ helicase provides a good example. In 2003, Lee et al. reported that phosphorylation of FANCJ (then known as BRCA1 Interacting C-terminal Helicase or BACH1) mediates its specific interaction with the carboxyl-terminal domain of the tumor suppressor BRCA1 [244]. This interaction was determined to be required for DNA damage-induced checkpoint control. In other work, FANCJ was found to be acetylated at its extreme carboxyl terminus which enhances DNA end-processing that is a prerequisite for HR repair and normal checkpoint maintenance [245]. RecQ DNA helicases, including WRN [246] and BLM [247], are also subject to post-translational modifications which regulate their pathway functions in the DNA damage response and DNA repair. Helicase proteins modified by ubiquitylating ligases, acetyl transferases, or kinases can become susceptible to protein degradation, which may afford a more sustained response to changes in the cellular environment (for review, see [248]). New experimental approaches to assess the effects of post-translational modifications at the SM level [249,250] provide a more refined methodology for characterizing their outcome compared to earlier studies which dealt with mixed populations of helicase molecules. Assessing the impact of post-translational modifications of helicase proteins at high-resolution appears to still be at its infancy. This field is likely to expand rapidly and provide interesting new insights into the regulation of helicase activity.
9. Pathway Functions of DNA Helicases
In this section we provide a historical account of discoveries pertaining to DNA helicases and their pathway functions, which serves as a backdrop for the subsequent section on the roles of DNA helicases in genetic diseases and cancer. Due to its vast nature, we have elected to limit the discussion to the functional roles of DNA helicases in the replication stress response or regulation of gene expression. The roles of DNA helicases in DNA repair pathways are discussed in reference [251] and referred to in other sections of this review, so we will not address these here.
9.1. Replication Stress
To our knowledge, the E. coli RecQ helicase was the first DNA helicase implicated in a cellular response to environmental manipulation that causes replication stress. In 1984 Hanawalt and colleagues reported that a recQ mutant strain was resistant to thymineless death and displayed increased sensitivity to UV light [252]. Just over ten years later, the first discovery of a RecQ helicase disorder was reported [181] (see Genetic Diseases section, Section 10). As Hanawalt would discuss in a 2015 perspective [253], this early discovery of RecQ’s relationship to thymineless death provided a proof-of-principle that research in unicellular organisms can yield great insight into human health. RecQ was indeed found to be a DNA helicase and genetic evidence suggested its involvement in HR repair [67]. RecQ is also thought to function in conjunction with structure-specific nucleases (e.g., RecJ) at replication forks blocked by UV light-induced photoproducts [254]. As it turns out, physical and functional interactions of RecQ helicase with DNA nucleases acting at specific DNA junctions has been a prevailing theme over the years (see Protein Interactions and reference [76]). The helicase core domain of the S. cerevisiae Sgs1 helicase is highly conserved with E. coli RecQ [255]. A role for Sgs1 in resolving DNA topological stress was suggested by its interaction with a topoisomerase (e.g., Top3) [255]. A year after its discovery, Sgs1 was reported to interact with topoisomerase II, and this interaction was shown to be required for faithful chromosome segregation [256]. Sgs1 was also found to be important in suppressing cellular aging and in preventing nucleolar fragmentation [257]. Around this time (1997), it was discovered that mutation of rqh1 (a Sgs1 homolog in S. pombe) compromised the ability of fission yeast to restart DNA synthesis after exposure to drugs that inhibit replication [258]. The Whitby laboratory showed that expression of a HJ resolvase in the rqh1 mutant strain partially rescued sensitivity to UV light or hydroxyurea (HU), suggesting the aberrant accumulation of recombinational DNA intermediates when Rqh1 was absent [259]. Restarting stalled forks due to topological stress or DNA damage appears to be the primary role of Sgs1, and by analogy Rqh1. Loss of Sgs1 (or its human homolog BLM, defective in BS (see below)) leads to stalled fork structures acted upon by structure-specific nucleases. These events may underlie the elevated sister chromatid exchange (SCE) observed in BLM-deficient cells [260].
The role of BLM in genome stability is likely to be more complex than what is observed for RecQ homologs in lower eukaryotes. For example, BLM was found to associate with BRCA1 and other DNA repair proteins implicated in MMR (MSH2, MSH6, MLH1), double-strand break signaling/repair (ATM, RAD50-MRE11-NBS1), and replication (PCNA, RFC) in a multi-protein complex known as BRCA1-associated genome surveillance complex (BASC). This complex has been proposed to coordinate multiple DNA transactions during replication of DNA containing damage or unusually folded secondary structure [261]. BLM was also shown to associate with the FANCJ helicase in response to replication stress, and the two DNA helicases physically and functionally interact [262]. The Hickson laboratory reported that BLM is phosphorylated in response to cellular HU exposure, and the phosphorylated form of BLM is required for recovery from S-phase arrest [263]. It will be of interest to ascertain the mechanism whereby BLM acts to stabilize stalled replication forks or to promote recovery from S-phase arrest. It seems likely to be related to a role played by S. cerevisiae Sgs1, which has been shown by chromatin immunoprecipitation (ChIP) assays to be associated with the ATM-related kinase Mec1 that serves to stabilize the replicative DNA polymerases alpha and epsilon at forks stalled by HU [264,265].
Sgs1 is likely to operate in at least a partially redundant role with non-RecQ DNA helicases including SRS2 (already implicated in the intra-S phase DNA damage checkpoint) and the 5’ to 3’ DNA helicase RRM3, as shown by synthetic lethality analyses by microarray (SLAM), a novel experimental approach at the time [266]. Since then, many papers describing synthetic lethal interactions of DNA helicases have appeared in the literature suggesting a general theme that helicases respond to replication stress via extended networks with numerous genetic interactions (for examples, see [267,268,269,270]). The recent identification of WRN helicase (mutated in Werner syndrome (WS), see below) as a synthetic lethal gene in microsatellite unstable cancers with defects in DNA MMR genes emphasizes the point [271,272,273,274]. The challenge in functional genomics remains to understand how, and in what context, certain DNA helicases have unique pathway functions. For example, experimental evidence suggests that Sgs1 acts to prevent the accumulation of Rad51-dependent cruciform DNA structures at damaged replication forks that have undergone incomplete maturation of recombination intermediates [275]. Although there is only one RecQ helicase in yeast, there are five in humans implying unique functions for each. Indeed, distinct genetic disorders are attributed to mutations in three of the five human RecQ helicases (see Genetic Diseases, Section 10). Yet there seems to be overlap in DNA substrate specificity and protein partners. How tasks are divided among the human RecQ helicases in pathway functions during the replication stress response remains an active area of investigation.
While much of the focus on RecQ helicases and replication stress has historically centered on Sgs1 and BLM, the human WRN helicase-nuclease appears to also play an important role. This notion was first suggested by Pichierri et al. (2001) who determined that cells from WS patients displayed poor recovery from exposure to camptothecin or HU, leading to elevated strand breaks [276]. A technical breakthrough was made by the Monnat laboratory in 2007 when they showed that cellular recovery from replication fork arrest was compromised upon acute depletion of WRN by RNA interference [277]. Subsequently, Pirzio et al. presented experimental data suggesting a catalytic requirement for the WRN helicase activity, but not its exonuclease activity, in maintaining fragile site stability by suppressing chromosomal breakage at fork stalling sites [278]. A technique-driven advance in characterizing the role of WRN in the replication stress response was made when Sidorova et al. utilized DNA fiber analysis to show that WRN depletion reduced fork elongation rate in cells exposed to HU or the base alkylating agent methylmethanesulfonate [279]. Collapsed replication forks caused by ICL damage-induced double strand breaks are also resolved by WRN, as shown by the Bohr laboratory [280]. In this case WRN was proposed to facilitate ATM activation and S-phase checkpoint. This is in contrast to WRN’s role in response to mild replication stress in which WRN was observed to regulate ATR-driven checkpoint activation [281].
Cell-based SM studies from the laboratories of Alessandro Vindigni and Massimo Lopes have begun to address the apparently unique but coordinated roles of RECQL1, DNA2, and WRN to restart replication forks that have been stalled by various pharmacological agents. DNA topoisomerase inhibition causes replication forks to reverse, and RECQL1 helps forks restart by promoting fork reversal of regressed forks in a manner that is negatively regulated by Poly(ADP)ribose polymerase (PARP) 1 [103] (Figure 3D). WRN and DNA2, on the other hand, are involved in nucleolytic processing of reversed forks [104] (Figure 3E). DNA2 nuclease activity and WRN ATPase/helicase activity together degrade forks with a 5’ to 3’ polarity, which promotes replication restart. In our own work, RECQL1’s catalytic strand separation activity was demonstrated to facilitate normal fork dynamics by governing RPA’s availability during replication stress [282]. In the latest advance (2019), Sharma’s laboratory characterized a checkpoint-dependent role of RECQL1 to respond to replication stress induced by the chemotherapy drug gemcitabine [283].
In the last several years, Pavel Janscak’s group and their collaborators have led the charge to characterize RECQL5’s role in helping cells cope with replication stress. Their results suggest that RECQL5 helps to prevent fork stalling in genes actively being transcribed by RNA Polymerase I or II by alleviating collisions between transcription and replication machinery [284]. By another mechanism, RECQL5 is proposed to collaborate with MUS81 endonuclease to process stalled replication forks at common fragile sites [285]. In addition, RECQL5 was reported to disrupt RAD51 filaments on stalled forks at common fragile sites, making the transition from fork stalling at R-loops to replication restart go smoothly [286].
Although the RecQ helicases have dominated the landscape for study of the replication stress response, helicases of other families have also been implicated. The Fe-S cluster helicase FANCJ represents a good example. Beyond FANCJ’s role in ICL repair and HR repair of double-strand breaks, Gong et al. reported that FANCJ interacts with TopBP1, a protein known to be important for the DNA replication checkpoint that helps cells respond to stalled forks [287]. FANCJ’s interaction with TopBP1 was shown to be required for Chk1 and RPA phosphorylation after cellular exposure to a replication stress-inducing agent, which in turn is necessary for the replication checkpoint. It is plausible that the chromosomal instability observed in FANCJ-deficient cells [262] is at least partially attributed to a defective replication checkpoint. More recent work solidifies the importance of FANCJ in maintaining the genomic stability of microsatellite DNA sequences during replication stress [288,289]. We have determined, using biochemical and genetic approaches, that a minimal threshold of FANCJ catalytic activity is required to suppress chromosomal DNA damage during cellular conditions of replication stress [290]. The Cantor laboratory has reported that a delicate balance between FANCJ and the fork remodeling factor HLTF is necessary to maintain fork stability [105] (Figure 3F).
DNA helicases like E. coli RecG [106] or human WRN [107,108] and BLM [107,109] may promote fork regression when a replication fork stalls to allow fork protection and eventual replication restart (Figure 3G). Although not considered bona fide DNA helicases per se, ATP hydrolysis-driven DNA translocases share many features in common with DNA helicases and play a prominent role in the response to replication stress. The first to be described was HARP, now more commonly designated SMARCAL1 (SWI/SNF-related matrix-associated, actin-dependent regulator of chromatin, subfamily-like A1). SMARCAL1 interacts with RPA and aids in replication fork restart [291,292]. SMARCAL1 performs its cellular functions of fork regression and HJ branch-migration to maintain genomic stability when forks encounter DNA damage [293]. In addition to SMARCAL1, the DNA translocases ZRANB3 and HLTF are involved in remodeling stalled fork structures as well (see [294] for review).
9.2. Transcriptional Regulation
As components of the general transcription factor TFIIH, the XPB and XPD helicases help to regulate gene expression by RNA polymerase II [295]. Egly and Coin offered an excellent review on the history of TFIIH for readers to learn more about this protein complex and its regulatory roles [295]. In addition to transcriptional activation, both the XPB and XPD helicases contribute to NER [251,296]. Missense mutations in XPB or XPD are linked to genetic diseases (see below), and both helicase subunits are essential in humans and other eukaryotes. XPB ATPase activity is required to locally open the duplex region in the vicinity of the transcriptional start site, as well as enable promoter escape, early elongation, and re-initiation of transcription when RNA polymerase II stalls [295]. Although molecular evidence has demonstrated that the catalytic DNA unwinding function of XPD is not required for its role in TFIIH transcriptional activation, the absence of XPD altogether destabilizes TFIIH which severely compromises transcription [297].
Both XPB and XPD are thought to play an additional role in transcriptional regulation that is related to their interactions with G4 DNA structures. G4 motifs are known to be enriched in promoter regions [298], suggesting that G-quadruplex structures residing within or downstream of promoters are utilized by the cell for transcriptional regulation. The Maizels laboratory pursued the hypothesis that the XPB and XPD helicases bind genomic G4 DNA structures, and the protein: DNA interactions are important for their transcriptional regulatory roles [98]. By ChIP Sequence analysis, they were able to show that 40% of the XPB and XPD binding sites overlapped with human DNA sequences predicted to form G4. Although a direct linkage of XPB or XPD G4 binding to transcriptional regulation was not shown, the authors determined that the S. acidocaldarius XPD helicase resolves a G4 DNA substrate in vitro. Further studies in this area are necessary for a mechanistic understanding of the importance of XPB or XPD interaction with promoter-localized G4 DNA in transcriptional regulation.
Of the five RecQ helicases, the strongest evidence for a primary role in regulation of RNA polymerase II transcription exists for RECQL5. The first report of RECQL5’s interaction with RNA polymerase II was in 2008, when its role in negatively regulating transcription was proposed [299]. RECQL5 was subsequently shown to suppress double strand breaks dependent on RNA polymerase II transcription [300]. In reconstitution experiments, RECQL5 was shown to inhibit both initiation and elongation by RNA polymerase II [301]. Domains in RECQL5 that interact with the initiation and elongation forms of RNA polymerase II were mapped [302]. Conversely, RECQL5 was shown to interact with a phosphorylated C-terminal domain of RNA polymerase II during transcription elongation [303]. Structural studies showed that RECQL5 binds to a site that overlaps with the binding site for the transcription factor TFIIS, positioning itself such that transcription elongation is sterically blocked [304]. An analysis of the genome-wide RNA polymerase II density profile suggested that RECQL5 acts to regulate movement of RNA polymerase II across genes in a manner that reflects transcription stress, where chromosomal breakpoints are localized [305]. An understanding of the basis for RECQL5’s modulatory effect on RNA polymerase II transcription began with the finding that the cellular topoisomerase TOPI is SUMO-lyated in a RECQL5-dependent manner, which prevents it from causing DNA damage at transcriptionally active chromatin [306]. Based on further studies in human cells, a model was proposed that RECQL5 helicase helps to prevent replication-transcription collisions [284]. A role for RECQL5 in restarting DNA replication after encounters with co-transcriptional R-loops and in collaboration with numerous other DNA repair factors was proposed recently [286].
Evidence suggests that G4-resolving or G4-interacting RecQ helicases influence gene expression. Brad Johnson’s laboratory provided the first evidence that cellular deficiency of the G4-resolving WRN and BLM helicases perturbs expression profiles of genes with G4-forming potential, as demonstrated by microarray analyses [307]. Further studies suggested that a deficiency in WRN [308] versus BLM [309] resulted in differential regulation of distinct sets of genes with enriched G4 motifs at the transcriptional start site or exon 1/intron 1 boundary, suggesting unique roles of the two helicases in gene expression. ChIP experiments suggested that RECQL1 preferentially binds to G4-forming genomic DNA sequences in a manner that is important for regulation of gene expression as well [310]. There is much yet to be learned regarding the role of DNA helicases in transcriptional regulation.
10. Genetic Diseases Characterized by Premature Aging and Cancer Linked to Molecular Defects in DNA Helicases
The decade of the 1990s marked an important era in helicase discovery as it relates to human disease. It was during this period that hereditary disorders characterized by DNA repair defects and/or chromosomal instability, a predisposition to cancer, and in certain cases, accelerated aging phenotypes were linked to mutations in helicase genes. Discovery of new rare hereditary disorders characterized by features of accelerated aging and/or cancer continues in the 21st Century. With a 2011 estimate of 31 nonredundant DNA helicases encoded by the human genome [14], it seems probable that more DNA helicase-linked genetic diseases will be identified, given the fundamentally important roles of ATP-dependent DNA unwinding enzymes in nucleic acid metabolism. However, as suggested in a recent review by Ray Monnat and colleagues, loss of function or compound haploinsufficiency for certain DNA helicase genes (e.g., RECQL1, RECQL5) may not yet be linked to a disease because their absence or dysfunction is incompatible with life (i.e., embryonic lethal) [311] or the rare causative mutation has not been identified. Genomics is still a young field and there are individuals worldwide with rare diseases in which the causative mutations have not yet been precisely mapped as challenges in the application of genome sequencing still exist [312]. In the following sections, we will describe several well-known genetic disorders attributed to mutations in DNA helicase genes from a historical perspective so the reader can appreciate the steady progress being made.
10.1. Xeroderma Pigmentosum, Cockayne Syndrome, and Trichothiodystrophy
In 1990, a helicase gene mutated in the skin cancer disease Xeroderma pigmentosum was identified by cDNA cloning and genetic complementation of the UV light sensitivity defect in UV5 Chinese hamster ovary cells [313]. This gene, originally designated ERCC2 and later dubbed XPD, was observed to share sequence similarity with the yeast RAD3 gene implicated in NER. Interestingly, XPD has roles in both DNA repair and transcription. Moreover, molecular defects in XPD are responsible for three distinct genetic disorders: Xeroderma pigmentosum, Xeroderma pigmentosum combined with Cockayne syndrome, and Trichothiodystrophy [314]. The purified XPD gene product was confirmed to be a DNA-dependent ATPase and DNA helicase in subsequent work [315]. XPD’s ATPase/helicase function are required for NER but not its role in transcription [297]. Based on collective evidence from the laboratories of Naegeli [316] and Hanaoka [317], it seems likely that XPD’s role in NER damage recognition is to scan the DNA molecule to verify the lesion (for review, see [318]).
For a comprehensive review of the multi-faceted roles of XPD in cellular nucleic acid metabolism, please see [319]. By virtue of its Fe-S cluster, XPD is capable of DNA-mediated redox signaling [320]. The Barton laboratory proposed a model in which DNA charge transport allows XPD and other redox active DNA repair proteins to efficiently search for DNA damage in the genome [321]. As discussed below, other human DNA helicase genes sharing sequence similarity in the helicase core domain with XPD are also implicated in hereditary disorders characterized by chromosomal instability and DNA repair defects.
Mutations in a second gene encoding a putative DNA helicase (originally designated ERCC3 and subsequently named XPB) were linked to Xeroderma pigmentosum and Cockayne syndrome in 1990 [322,323]. XPB mutations are also implicated in Xeroderma pigmentosum with neurological abnormalities and Trichothiodystrophy [324]. As mentioned above, XPB (like XPD) is involved in both NER and transcription.
10.2. Bloom Syndrome
For many years it was known that the hypermutability associated with BS was accompanied by replication defects and elevated SCE, the latter serving as a clinical diagnostic test for the disorder [325]. In 1995, Ellis, German and colleagues utilized a classical mapping technique known as positional cloning to determine the location and identity of bi-allelic BS-causing mutations in a novel gene (designated BLM) sharing sequence similarity with the coding region for the ATPase/helicase core domain of the bacterial RecQ helicase [181]. This discovery was not only exciting for scientists interested in aging, but also those studying cancer biology. Individuals with BS display a broad spectrum of blood and solid-tumor cancers as well as immuno-deficiency, short stature, and certain features of rapid aging. The apparently contrasting roles of the BLM helicase to either promote or dissuade recombination has made it difficult to grasp the molecular basis for the elevated SCE observed in BS. However, biochemical studies from the Hickson laboratory using oligonucleotide-based or plasmid-based double HJ substrates suggests that BLM has a unique role among the RecQ helicases. BLM collaborates with a DNA topoisomerase to dissolve the complex structure which is a presumed intermediate of HR or converging replication forks [326].
In addition to its role in modulating recombinational DNA repair by branch-migrating mobile displacement (D)-loop substrates [327] or acting upon HJ structures [63], experimental evidence suggests that BLM plays a role in DNA end-resection, a process that is required to generate a single-strand overhang for strand invasion into homologous duplex DNA. From in vitro reconstitution experiments, BLM was shown to act in complex with the structure-specific helicase-nuclease DNA2 or EXO1 to perform long strand resection [328,329]. BLM has also been implicated in protection and repair of replication forks [330] and resolution of ultra-fine anaphase DNA bridges [331]. However, it is still unclear how defects in these BLM-related activities are responsible for the cellular and clinical features of BS. Presumably, BLM helicase has multiple roles in DNA transactions in vivo and its molecular deficiency contributes to chromosomal instability in BS through several avenues [332].
10.3. Werner Syndrome
The discovery of the BLM gene was rapidly followed in the subsequent year (1996) with a report by Yu et al. detailing the positional cloning of the gene mutated in the premature aging disorder WS [333]. The WRN gene, defective in WS, also encodes a DNA helicase of the RecQ family. Interestingly, a conserved exonuclease domain, characteristic of proofreading nucleases, was also identified in WRN [334] making it unique among the five human RecQ helicases. WS is distinct from BS in that it much more dramatically displays many of the clinical features of aging early in life. The clinical features appear after adolescence, typically in the mid to late 20’s, in an affected person. A particularly remarkable aspect of WS is that practically all of the symptoms of normal aging, including diabetes, osteoporosis, cataracts, heart disease, wrinkled skin and gray hair, manifest early in life and the average lifespan is 46 years. Neurological decline is generally not thought to be a symptom of WS. However, due to the premature death of WS patients and the limited number of cases in the WS registry, the prevalence of clinical features resembling early Alzheimer’s Disease or dementia in some WS patients cannot be excluded. See [335] for a recent discussion of WS and the central nervous system. A clinical feature that distinguishes WS from BS is that cancer types in WS are less broad than in BS. Soft tissue sarcomas and other neoplasms are prevalent in WS, whereas BS displays a broad spectrum of cancers including solid tumor and blood cancers [336].
From a historical perspective, a significant advance in understanding the molecular pathology of WS was made by discoveries linking WRN defects to abnormal telomere metabolism. This began with mouse model studies showing that co-deficiency of WRN and the telomerase RNA-protein complex in later generation mice with short telomeres displayed accelerated aging and chromosomal instability as is typically observed in human WS patients [337,338,339]. Furthermore, human cells defective in WRN helicase activity were found to display defective lagging strand synthesis in the telomeres [340]. Following these advances, biochemical characterization of the WRN protein revealed a DNA substrate specificity and protein interactions that suggested a specialized molecular role in telomere metabolism [234]. More general roles of WRN in replication and recombination are also likely to come into play. Despite the work of many laboratories and over two decades of research, the molecular basis for WS remains enigmatic. Perhaps novel approaches that examine the crosstalk of DNA damage with cellular metabolism and senescence observed in other forms of progeria (e.g., [341]) and a focus on why WS is an adult-onset disorder [342] may help to move the field forward.
10.4. Rothmund-Thomson Syndrome and Related RECQL4 Disorders
In 1999 Rothmund-Thomson syndrome (RTS) was shown to be linked to bi-allelic mutations in a third member of the RecQ family, the RECQL4 helicase [343]. Further studies revealed that RECQL4 mutations are linked to not only RTS, but also Baller-Gerold syndrome [344] and RAPADILINO syndrome [345], all characterized by stunted growth, radial ray defects, and skeletal abnormalities. The molecular basis for the complex disease symptoms associated with RECQL4 mutations remains to be determined, but the recent structure of RECQL4 may help to explain the defects caused by some of the mutations [198]. Of the five human RecQ helicases, RECQL4 appears to have the weakest ATP-dependent DNA unwinding function in vitro [346,347]. Like the other RecQ helicases it can also perform strand annealing [348], the opposite of DNA unwinding. Adding to the complexity, recent work using mouse models suggests that RECQL4 helicase activity is not required for normal development or physiological functions [349]. Consequently, it remains unclear if defective RECQL4 helicase activity in humans per se is implicated in any of the three RECQL4-linked genetic disorders. Consistent with this hypothesis, the essential role of RECQL4 in hematopoiesis, as shown in RECQL4-deficient mice, was found not to be dependent on its helicase activity [350], leaving the importance or role of its modest DNA unwinding activity unexplained.
10.5. Fanconi Anemia
As alluded to above, a new cadre of clinically relevant diseases have been identified in which the mutated genes all encode proteins which share sequence similarity within the helicase core domain of XPD. Remarkably, in 2005 three research teams independently discovered that mutations in a DNA helicase gene (designated FANCJ and sharing sequence similarity with XPD) were implicated in a genetically complex disorder known as FA characterized by congenital abnormalities, progressive bone marrow failure, and cancer [351,352,353]. A peculiar feature of the XPD/FANCJ helicase family is the presence of an Fe-S cluster [354] that serves a critical structural and biochemical function most likely as a molecular wedge in helicase-catalyzed duplex DNA unwinding [192,193,354]. Consistent with these findings, a pathogenic amino acid substitution in the Fe-S domain of FANCJ was shown to abolish its DNA repair function in vivo and inactivate its helicase function in vitro [355]. Cells from individuals carrying bi-allelic FANCJ mutations, like the mutations in other FA genes, are hypersensitive to DNA cross-linking agents. It is believed that FANCJ’s role in HR repair is critical for healing of double-strand breaks associated with interstrand cross-linking (ICL)-induced DNA damage in replicating cells (for review, see [356]). However, FANCJ’s molecular mechanism of action remains poorly understood. In addition, FANCJ plays an even broader role in the cellular response to replication stress [105,290]. It was recently shown by biochemical and genetic studies that a greater threshold of FANCJ helicase activity is required for a robust response to ICL-induced damage compared to double-strand breaks induced by the chemical bleomycin or replication stress caused by the DNA polymerase inhibitor aphidicolin or G-quadruplex ligand telomestatin [290]. With emerging evidence that endogenous macromolecule damage induced by formaldehyde and aldehyde derivatives may be the causative force underlying FA [357,358], a central challenge will be to determine how molecular defects in FANCJ and the 20+ other proteins implicated in the FA pathway are responsible for the characteristic disease outcomes, i.e., accelerated decline of the hematopoietic stem cell compartment and other features of aging [359].
In addition to FANCJ, a second DNA-dependent ATPase designated FANCM was discovered and bi-allelic mutations were originally thought to be linked to FA [360]. However, this turned out not to be the case as that individual harbored bi-allelic mutations in the FANCA gene [361], which was already shown to be genetically linked to FA [362]. More recent work implicates FANCM as a facilitator of HR and replication by collaborating with a number of genome caretaker proteins. Consistent with this proposal, FANCM is mutated in various cancers and suspected to act globally as a tumor suppressor (for review, see [363]). From a biochemical standpoint, FANCM was shown in 2005 by Meetai et al. to possess a dsDNA translocase activity. However, FANCM is not a bona fide DNA helicase capable of catalytically separating complementary strands of a duplex DNA substrate [360]. Further studies revealed that FANCM is capable of branch-migrating HJs and certain DNA structures associated with stalled replication forks [364,365,366,367]. FANCM’s branch-migration activity may be relevant to its collaborative role with the BLM helicase to suppress SCEs and confer resistance to DNA cross-linking agents [368]. Using an elegant SM technique to visualize replication forks as they encounter ICLs, Seidman’s laboratory provided definitive evidence that FANCM’s ATPase/translocase activity facilitates replication traverse of the ICL [369]. The molecular mechanism(s) of ICL tolerance and repair continues to advance rapidly with novel experimental strategies and model systems; the reader is referred to several recent reviews on this topic [370,371,372,373].
10.6. Dyskeratosis Congenita and Warsaw Breakage Syndrome
The RTEL1 helicase, mutated in Dyskeratosis congenita (DC) or its more severe form Hoyeraal-Hreidarsson syndrome (HHS), and the DDX11 helicase mutated in Warsaw Breakage syndrome (WBS) have most recently joined the group of clinically relevant Fe-S DNA helicases. Both diseases are chromosome instability disorders, but unique in their presentation. Cells from WBS individuals display elevated sister chromatid cohesion defects [374], whereas DC or HHS, due to RTEL1 mutation, is characterized by telomere shortening [375,376,377]. Evidence suggests that RTEL1 plays a role in the processing of telomeric D-loops (so-called t-loops), resolution of telomere-associated G-quadruplex DNA [378], and regulation of HR repair [379]. Until only very recently, definitive molecular evidence for RTEL1’s precise role(s) was lacking. However, new studies by Sarek et al. demonstrate that RTEL1’s access to t-loops to enable smooth telomere replication during the cell cycle is governed by the phosphorylation state of the shelterin protein TRF2 [380]. A more general role for RTEL1, acting together with PCNA in replication of the whole genome, has also been suggested based on DNA fiber studies of mouse embryonic fibroblasts from RTEL1 mutant mice [123].
Interest in the biology of DDX11 helicase continues to grow with the identification of new patients [381], development of cell-based models [382,383,384,385], and an increased understanding of the molecular events important for sister chromatid cohesion [386,387,388]. However, compared to the Fe-S helicases XPD, FANCJ, and the human RecQ helicases, the mechanism and functional importance of helicase-catalyzed DNA unwinding by DDX11 (or RTEL1) has been largely under-studied. Research from the Pisani laboratory suggests that the interaction of DDX11 with the replication fork-protection factor Timeless is important for fork recovery from replication stress and promotion of sister chromatid cohesion [383,384]. A role of Timeless as a checkpoint protein that couples cell cycle progression with circadian rhythm [389,390] raises the question if DDX11 or other DNA helicases implicated in the DNA damage response and genome maintenance are involved in the cell-autonomous clock that regulates physiologic functions.
10.7. Mitochondrial Twinkle Helicase Diseases
Hereditary mitochondrial diseases attributed to mutations in the human nuclear-encoded mitochondrial replicative helicase Twinkle (C10orf2) include progressive external ophthalmoplegia (PEO), ataxia-neuropathy syndromes (e.g., infantile-onset spinocerebellar ataxia), and mitochondrial DNA depletion syndromes (e.g., hepatocerebral syndrome) (for review, see [391,392]). The genetics of Twinkle-linked mitochondrial disorders is complex, and often the inheritance is attributed to an autosomal dominant mutation. The Copeland, Falkenberg, and Spelbrink laboratories have undertaken efforts to characterize a number of clinically relevant Twinkle mutations and their biochemical effects, which suggest partial reduction in activity, destabilization of the protein, and interference in oligomerization [393,394,395]. The Twinkle gene was first discovered in 2001 by Spelbrink et al. [396]. In this work, they identified Twinkle mutations linked to adult onset PEO characterized by multiple mitochondrial deletions. A number of groups have been involved in the biochemical and biophysical characterization of Twinkle which, like other replicative helicases, forms multi-subunit (6 or 7-membered) rings [397,398]. A transgenic mouse model for a Twinkle mutation was found to recapitulate the PEO characteristic of the human disorder [399].
11. Helicase Mutations and Predisposition to Cancer
Although the genetic linkage of DNA helicase defects to rare chromosomal instability disorders has been emphasized in this section, it should be stated that there are numerous studies documenting mutations in DNA helicase genes as being associated with cancers [400]. For example, a recent study established that rare missense alleles of BRIP1/FANCJ confer risk for breast as well as ovarian cancer [401]. This work is consistent with a previous study that truncating mutations in BRIP1/FANCJ confer susceptibility to breast cancer [402]. The association of helicase mutations with various cancers, as well as hereditary diseases with a cancer predisposition, is believed to reflect the vast and important roles of DNA helicases in DNA repair, maintenance of genomic stability, and cellular checkpoint responses important for chromosomal stability. Thus, DNA helicases belong to a class of genome caretaker proteins important for cancer suppression.
12. Small Molecule Modulation of DNA Helicases
We have discussed how helicase function can be modulated by mutation, post-translational modification, or protein interaction. Yet another form of helicase regulation can be achieved by pharmacological modulation with helicase-interacting small molecules. This field of study was motivated in large part by earlier reports that DNA repair proteins (e.g., (PARP)) are inhibited by specific small molecules (e.g., olaparib in the case of PARP-1) which can be used to modulate DNA repair capacity in human cells, particularly in defined genetic backgrounds (BRCA) [403,404]. Such an approach is postulated to be a new methodology for anti-cancer therapy, and PARP inhibitors are being used in the clinic (e.g., olaparib [405], rucaparib [406]) or in clinical trials. Other DNA repair inhibitors are currently being investigated for their vulnerability to pharmacological inhibition by small molecules that interact with their targets [407].
The first reported class of helicase inhibitors were targeted against viral helicases in an effort to suppress viral diseases, dating back to the early 2000’s (for review, see [408]). The first described human helicase inhibitor was directed against WRN [409], followed by a BLM helicase inhibitor [410]. These compounds were found to be bioactive with human cells grown in culture in a helicase target-dependent manner, suggesting their specificity. A second WRN helicase inhibitor, structurally related to the first, was found to be effective in sensitizing FA mutant cells to very low concentrations of an ICL-inducing agent [411], suggesting that the WRN-catalyzed DNA unwinding function helps cells cope with ICL-induced DNA damage possibly by a replication stress response or an HR-dependent mechanism. More recently discovered small molecule inhibitors directed against helicase proteins such as the human DNA2 helicase-nuclease [412,413], human immunodeficiency virus (HIV) DDX3 RNA helicase [414,415], and the eukaryotic MCM replicative helicase [416,417,418] have been described. Important advances for the development of helicase inhibitors are likely to come with organismal genetic models (e.g., mice) and successful virtual screening to develop more potent and specific helicase inhibitors [419]. Although still far from clinical application, small molecule helicase inhibitors have already proven to be useful tools in basic science research.
13. Summary
In this review we have provided a historical perspective of the study of DNA helicases by highlighting some of the seminal discoveries for this unique class of nucleic acid metabolizing enzymes. Our chronological account is meant to recognize key findings that have helped to shape this fertile area of study and provide a resource for early career nucleic acid biologists. In addition, we hope that this review will inspire researchers to continue their pursuit of new directions for investigation of DNA helicases in the years to come. While RNA helicases were not discussed in this review, we recognize that these enzymes are equally tantalizing in their interest and impact.
Author Contributions
R.M.B.J. and S.W.M. equally contributed to the conceptualization, writing, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the Intramural Research Program, National Institute on Aging, National Institutes of Health (R.M.B.J.).
Acknowledgments
We thank Marc Raley at NIA Visual Media Services for the artwork. We are grateful to the many helicase researchers and their dedication to scientific investigation. Because of the vast nature of DNA helicase literature, we were unable to cover all topics worthy of consideration. We apologize to those authors whose work we did not discuss due to space constraints.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abdel-Monem, M.; Durwald, H.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 2. Chain separation by an ATP-dependent DNA unwinding enzyme. Eur. J. Biochem. 1976, 65, 441–449. [Google Scholar] [CrossRef]
- Abdel-Monem, M.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 1. Purification and characterization of a DNA-Dependent ATPase from Escherichia coli. Eur. J. Biochem. 1976, 65, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mackay, V.; Linn, S. Selective inhibition of the DNase activity of the recBC enzyme by the DNA binding protein from Escherichia coli. J. Biol. Chem. 1976, 251, 3716–3719. [Google Scholar] [PubMed]
- Lohman, T.M.; Fazio, N.T. How does a helicase unwind DNA? Insights from RecBCD Helicase. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1800009. [Google Scholar] [CrossRef] [PubMed]
- Geider, K.; Berthold, V.; Abdel-Monem, M.; Hoffman-Berling, H. The Single-Stranded DNA Phages; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1978; pp. 379–387. [Google Scholar]
- Kuhn, B.; Abdel-Monem, M.; Hoffmann-Berling, H. DNA helicases. Cold Spring Harb. Symp. Quant. Biol. 1979, 43 Pt 1, 63–67. [Google Scholar] [CrossRef]
- Kuhn, B.; Abdel-Monem, M.; Krell, H.; Hoffmann-Berling, H. Evidence for two mechanisms for DNA unwinding catalyzed by DNA helicases. J. Biol. Chem. 1979, 254, 11343–11350. [Google Scholar]
- Seki, M.; Enomoto, T.; Hanaoka, F.; Yamada, M. DNA-Dependent adenosinetriphosphatase B from mouse FM3A cells has DNA helicase activity. Biochemistry 1987, 26, 2924–2928. [Google Scholar] [CrossRef]
- Seki, M.; Enomoto, T.; Yanagisawa, J.; Hanaoka, F.; Ui, M. Further characterization of DNA helicase activity of mouse DNA-dependent adenosinetriphosphatase B (DNA helicase B). Biochemistry 1988, 27, 1766–1771. [Google Scholar] [CrossRef]
- Yanagisawa, J.; Seki, M.; Kohda, T.; Enomoto, T.; Ui, M. DNA-Dependent adenosinetriphosphatase C1 from mouse FM3A cells has DNA helicase activity. J. Biol. Chem. 1992, 267, 3644–3649. [Google Scholar]
- Tuteja, N.; Tuteja, R.; Rahman, K.; Kang, L.Y.; Falaschi, A. A DNA helicase from human cells. Nucleic Acids Res. 1990, 18, 6785–6792. [Google Scholar] [CrossRef] [Green Version]
- Hehman, G.L.; Hauswirth, W.W. DNA helicase from mammalian mitochondria. Proc. Natl. Acad. Sci. USA 1992, 89, 8562–8566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuteja, N.; Phan, T.N.; Tewari, K.K. Purification and characterization of a DNA helicase from pea chloroplast that translocates in the 3’-to-5’ direction. Eur. J. Biochem. 1996, 238, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Umate, P.; Tuteja, N.; Tuteja, R. Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol. 2011, 4, 118–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuteja, N.; Tuteja, R. Prokaryotic and eukaryotic DNA helicases. Essential molecular motor proteins for cellular machinery. Eur. J. Biochem. 2004, 271, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Bessman, M.J.; Lehman, I.R.; Simms, E.S.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. II. General properties of the reaction. J. Biol. Chem. 1958, 233, 171–177. [Google Scholar] [PubMed]
- Lehman, I.R.; Bessman, M.J.; Simms, E.S.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem. 1958, 233, 163–170. [Google Scholar]
- Hotta, Y.; Stern, H. DNA unwinding protein from meiotic cells of Lilium. Biochemistry 1978, 17, 1872–1880. [Google Scholar] [CrossRef]
- Venkatesan, M.; Silver, L.L.; Nossal, N.G. Bacteriophage T4 gene 41 protein, required for the synthesis of RNA primers, is also a DNA helicase. J. Biol. Chem. 1982, 257, 12426–12434. [Google Scholar]
- Hubscher, U.; Stalder, H.P. Mammalian DNA helicase. Nucleic Acids Res. 1985, 13, 5471–5483. [Google Scholar] [CrossRef] [Green Version]
- Stahl, H.; Droge, P.; Knippers, R. DNA helicase activity of SV40 large tumor antigen. EMBO J. 1986, 5, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Ryu, B.H.; Sugino, T.; Naumovski, L.; Friedberg, E.C. A new DNA-Dependent ATPase which stimulates yeast DNA polymerase I and has DNA-unwinding activity. J. Biol. Chem. 1986, 261, 11744–11750. [Google Scholar] [PubMed]
- Tuteja, R.; Malhotra, P.; Song, P.; Tuteja, N.; Chauhan, V.S. Isolation and characterization of an eIF-4A homologue from Plasmodium cynomolgi. Mol. Biochem. Parasitol. 2002, 124, 79–83. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-Subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. A conserved NTP-motif in putative helicases. Nature 1988, 333, 22. [Google Scholar] [CrossRef]
- Hodgman, T.C. A new superfamily of replicative proteins. Nature 1988, 333, 22–23. [Google Scholar] [CrossRef]
- Lane, D. Enlarged family of putative helicases. Nature 1988, 334, 478. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. A novel superfamily of nucleoside triphosphate-binding motif containing proteins which are probably involved in duplex unwinding in DNA and RNA replication and recombination. FEBS Lett. 1988, 235, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Koonin, E.V. Helicases: Amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 1993, 3, 419–429. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Higgins, D.; Prakash, L.; Prakash, S. Mutation of lysine-48 to arginine in the yeast RAD3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. EMBO J. 1988, 7, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
- Zavitz, K.H.; Marians, K.J. ATPase-Deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J. Biol. Chem. 1992, 267, 6933–6940. [Google Scholar] [PubMed]
- George, J.W.; Brosh, R.M., Jr.; Matson, S.W. A dominant negative allele of the Escherichia coli uvrD gene encoding DNA helicase II. A biochemical and genetic characterization. J. Mol. Biol. 1994, 235, 424–435. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Matson, S.W. A partially functional DNA helicase II mutant defective in forming stable binary complexes with ATP and DNA. A role for helicase motif III. J. Biol. Chem. 1996, 271, 25360–25368. [Google Scholar] [CrossRef] [Green Version]
- Dillingham, M.S.; Soultanas, P.; Wigley, D.B. Site-directed mutagenesis of motif III in PcrA helicase reveals a role in coupling ATP hydrolysis to strand separation. Nucleic Acids Res. 1999, 27, 3310–3317. [Google Scholar] [CrossRef] [Green Version]
- Zittel, M.C.; Keck, J.L. Coupling DNA-Binding and ATP hydrolysis in Escherichia coli RecQ: Role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 2005, 33, 6982–6991. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, B.; Keck, J.L. Grip it and rip it: Structural mechanisms of DNA helicase substrate binding and unwinding. Protein Sci. A Publ. Protein Soc. 2014, 23, 1498–1507. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.C.; Matson, S.W. Helicase motifs: The engine that powers DNA unwinding. Mol. Microbiol. 1999, 34, 867–877. [Google Scholar] [CrossRef]
- Tuteja, N.; Tuteja, R. Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. 2004, 271, 1849–1863. [Google Scholar] [CrossRef]
- Matson, S.W.; Tabor, S.; Richardson, C.C. The gene 4 protein of bacteriophage T7. Characterization of helicase activity. J. Biol. Chem. 1983, 258, 14017–14024. [Google Scholar] [PubMed]
- Bjornson, K.P.; Amaratunga, M.; Moore, K.J.; Lohman, T.M. Single-turnover kinetics of helicase-catalyzed DNA unwinding monitored continuously by fluorescence energy transfer. Biochemistry 1994, 33, 14306–14316. [Google Scholar] [CrossRef] [PubMed]
- Dessinges, M.N.; Lionnet, T.; Xi, X.G.; Bensimon, D.; Croquette, V. Single-molecule assay reveals strand switching and enhanced processivity of UvrD. Proc. Natl. Acad. Sci. USA 2004, 101, 6439–6444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstock, M.J.; Whitley, K.D.; Jia, H.; Sokoloski, J.; Lohman, T.M.; Ha, T.; Chemla, Y.R. Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science 2015, 348, 352–354. [Google Scholar] [CrossRef] [Green Version]
- Whitley, K.D.; Comstock, M.J.; Chemla, Y.R. High-resolution “Fleezers”: Dual-trap optical tweezers combined with single-molecule fluorescence detection. Methods Mol. Biol. 2017, 1486, 183–256. [Google Scholar] [CrossRef] [Green Version]
- Matson, S.W.; Kaiser-Rogers, K.A. DNA helicases. Annu. Rev. Biochem. 1990, 59, 289–329. [Google Scholar] [CrossRef]
- Yarranton, G.T.; Gefter, M.L. Enzyme-catalyzed DNA unwinding: Studies on Escherichia coli rep protein. Proc. Natl. Acad. Sci. USA 1979, 76, 1658–1662. [Google Scholar] [CrossRef] [Green Version]
- Matson, S.W. Escherichia coli helicase II (urvD gene product) translocates unidirectionally in a 3’ to 5’ direction. J. Biol. Chem. 1986, 261, 10169–10175. [Google Scholar]
- Subramanya, H.S.; Bird, L.E.; Brannigan, J.A.; Wigley, D.B. Crystal structure of a DExx box DNA helicase. Nature 1996, 384, 379–383. [Google Scholar] [CrossRef]
- Velankar, S.S.; Soultanas, P.; Dillingham, M.S.; Subramanya, H.S.; Wigley, D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 1999, 97, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Korolev, S.; Hsieh, J.; Gauss, G.H.; Lohman, T.M.; Waksman, G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-Stranded DNA and ADP. Cell 1997, 90, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Saikrishnan, K.; Powell, B.; Cook, N.J.; Webb, M.R.; Wigley, D.B. Mechanistic basis of 5’-3’ translocation in SF1B helicases. Cell 2009, 137, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuper, J.; Wolski, S.C.; Michels, G.; Kisker, C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012, 31, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, R.A.; Wu, C.G.; Spies, M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2012, 31, 503–514. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-loops as cellular regulators and genomic threats. Molecular Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Matson, S.W. Escherichia coli DNA helicase II (uvrD gene product) catalyzes the unwinding of DNA.RNA hybrids in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 4430–4434. [Google Scholar] [CrossRef] [Green Version]
- Matson, S.W.; George, J.W. DNA helicase II of Escherichia coli. Characterization of the single-stranded DNA-Dependent NTPase and helicase activities. J. Biol. Chem. 1987, 262, 2066–2076. [Google Scholar]
- Shin, J.H.; Kelman, Z. The replicative helicases of bacteria, archaea, and eukarya can unwind RNA-DNA hybrid substrates. J. Biol. Chem. 2006, 281, 26914–26921. [Google Scholar] [CrossRef]
- Lloyd, R.G.; Benson, F.E.; Shurvinton, C.E. Effect of ruv mutations on recombination and DNA repair in Escherichia coli K12. Mol. Gen. Genet. 1984, 194, 303–309. [Google Scholar] [CrossRef]
- Connolly, B.; Parsons, C.A.; Benson, F.E.; Dunderdale, H.J.; Sharples, G.J.; Lloyd, R.G.; West, S.C. Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc. Natl. Acad. Sci. USA 1991, 88, 6063–6067. [Google Scholar] [CrossRef] [Green Version]
- Parsons, C.A.; Tsaneva, I.; Lloyd, R.G.; West, S.C. Interaction of Escherichia coli RuvA and RuvB proteins with synthetic Holliday junctions. Proc. Natl. Acad. Sci. USA 1992, 89, 5452–5456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karow, J.K.; Constantinou, A.; Li, J.L.; West, S.C.; Hickson, I.D. The Bloom′s syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl. Acad. Sci. USA 2000, 97, 6504–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, A.; Tarsounas, M.; Karow, J.K.; Brosh, R.M.; Bohr, V.A.; Hickson, I.D.; West, S.C. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom′s and Werner′s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, A.S.; Tahmaseb, K.; Compton, S.A.; Matson, S.W. Resolving Holliday junctions with Escherichia coli UvrD helicase. J. Biol. Chem. 2012, 287, 8126–8134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezu, K.; Nakayama, K.; Nakayama, H. Escherichia coli RecQ protein is a DNA helicase. Proc. Natl. Acad. Sci. USA 1990, 87, 5363–5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runyon, G.T.; Bear, D.G.; Lohman, T.M. Escherichia coli helicase II (UvrD) protein initiates DNA unwinding at nicks and blunt ends. Proc. Natl. Acad. Sci. USA 1990, 87, 6383–6387. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr.; Matson, S.W. A point mutation in Escherichia coli DNA helicase II renders the enzyme nonfunctional in two DNA repair pathways. Evidence for initiation of unwinding from a nick in vivo. J. Biol. Chem. 1997, 272, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Grilley, M.; Griffith, J.; Modrich, P. Bidirectional excision in methyl-directed mismatch repair. J. Biol. Chem. 1993, 268, 11830–11837. [Google Scholar]
- Orren, D.K.; Selby, C.P.; Hearst, J.E.; Sancar, A. Post-incision steps of nucleotide excision repair in Escherichia coli. Disassembly of the UvrBC-DNA complex by helicase II and DNA polymerase I. J. Biol. Chem. 1992, 267, 780–788. [Google Scholar]
- Brosh, R.M., Jr.; Waheed, J.; Sommers, J.A. Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J. Biol. Chem. 2002, 277, 23236–23245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugreev, D.V.; Yu, X.; Egelman, E.H.; Mazin, A.V. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007, 21, 3085–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Sommers, J.A.; Wu, L.; Bohr, V.A.; Hickson, I.D.; Brosh, R.M., Jr. Stimulation of Flap endonuclease-1 by the Bloom’s syndrome protein. J. Biol. Chem. 2004, 279, 9847–9856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Sommers, J.A.; Choudhary, S.; Faulkner, J.K.; Cui, S.; Andreoli, L.; Muzzolini, L.; Vindigni, A.; Brosh, R.M., Jr. Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J. Biol. Chem. 2005, 280, 28072–28084. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sommers, J.A.; Brosh, R.M., Jr. Processing of DNA replication and repair intermediates by the concerted action of RecQ helicases and Rad2 structure-specific nucleases. Protein Pept. Lett. 2008, 15, 89–102. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Estep, K.N.; Butler, T.J.; Ding, J.; Brosh, R.M., Jr. G4-interacting DNA helicases and polymerases: Potential therapeutic targets. Curr. Med. Chem. 2019, 26, 2881–2897. [Google Scholar] [CrossRef]
- Mendoza, O.; Bourdoncle, A.; Boule, J.B.; Brosh, R.M., Jr.; Mergny, J.L. G-Quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [Green Version]
- Cheung, I.; Schertzer, M.; Rose, A.; Lansdorp, P.M. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-Rich DNA. Nat. Genet. 2002, 31, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-Quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-Specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Shin-ya, K.; Brosh, R.M., Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-Quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-Quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shin-ya, K.; Niedzwiedz, W. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J. Cell Biol. 2013, 201, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Lansdorp, P.; van Wietmarschen, N. Helicases FANCJ, RTEL1 and BLM act on guanine quadruplex DNA in vivo. Genes 2019, 10, 870. [Google Scholar] [CrossRef] [Green Version]
- Bochman, M.L.; Judge, C.P.; Zakian, V.A. The Pif1 family in prokaryotes: What are our helicases doing in your bacteria? Mol. Biol. Cell 2011, 22, 1955–1959. [Google Scholar] [CrossRef]
- Byrd, A.K.; Bell, M.R.; Raney, K.D. Pif1 helicase unfolding of G-quadruplex DNA is highly dependent on sequence and reaction conditions. J. Biol. Chem. 2018, 293, 17792–17802. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-Quadruplex motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Piazza, A.; Boule, J.B.; Lopes, J.; Mingo, K.; Largy, E.; Teulade-Fichou, M.P.; Nicolas, A. Genetic instability triggered by G-Quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res. 2010, 38, 4337–4348. [Google Scholar] [CrossRef] [Green Version]
- Ribeyre, C.; Lopes, J.; Boule, J.B.; Piazza, A.; Guedin, A.; Zakian, V.A.; Mergny, J.L.; Nicolas, A. The yeast Pif1 helicase prevents genomic instability caused by G-Quadruplex-Forming CEB1 sequences in vivo. PLoS Genet. 2009, 5, e1000475. [Google Scholar] [CrossRef] [Green Version]
- Sanders, C.M. Human Pif1 helicase is a G-Quadruplex DNA-binding protein with G-Quadruplex DNA-unwinding activity. Biochem. J. 2010, 430, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, M.A.; Singh, S.P.; Burgers, P.M.; Galletto, R. Complementary roles of Pif1 helicase and single stranded DNA binding proteins in stimulating DNA replication through G-Quadruplexes. Nucleic Acids Res. 2019, 47, 8595–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallgren, M.; Mohammad, J.B.; Yan, K.P.; Pourbozorgi-Langroudi, P.; Ebrahimi, M.; Sabouri, N. G-Rich telomeric and ribosomal DNA sequences from the fission yeast genome form stable G-Quadruplex DNA structures in vitro and are unwound by the Pfh1 DNA helicase. Nucleic Acids Res. 2016, 44, 6213–6231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA patrolling underlies diverse functions of Pif1 on R-Loops and G-Rich DNA. eLife 2014, 3, e02190. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA replication through G-Quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 2011, 145, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Dahan, D.; Tsirkas, I.; Dovrat, D.; Sparks, M.A.; Singh, S.P.; Galletto, R.; Aharoni, A. Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 2018, 46, 11847–11857. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; von Kobbe, C.; Sommers, J.A.; Karmakar, P.; Opresko, P.L.; Piotrowski, J.; Dianova, I.; Dianov, G.L.; Bohr, V.A. Werner syndrome protein interacts with human Flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001, 20, 5791–5801. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Otterlei, M.; Sommers, J.A.; Driscoll, H.C.; Dianov, G.L.; Kao, H.I.; Bambara, R.A.; Brosh, R.M., Jr. WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol. Biol. Cell 2004, 15, 734–750. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sommers, J.A.; Brosh, R.M., Jr. In vivo function of the conserved non-catalytic domain of Werner syndrome helicase in DNA replication. Hum. Mol. Genet. 2004, 13, 2247–2261. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bambara, R.A. Human Bloom protein stimulates Flap endonuclease 1 activity by resolving DNA secondary structure. J. Biol. Chem. 2005, 280, 5391–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Cong, K.; Panzarino, N.J.; Nayak, S.; Calvo, J.; Deng, B.; Zhu, L.J.; Morocz, M.; Hegedus, L.; Haracska, L.; et al. Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep. 2018, 24, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, P.; Lloyd, R.G.; Marians, K.J. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. USA 2001, 98, 8235–8240. [Google Scholar] [CrossRef] [Green Version]
- Machwe, A.; Xiao, L.; Groden, J.; Orren, D.K. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 2006, 45, 13939–13946. [Google Scholar] [CrossRef]
- Machwe, A.; Xiao, L.; Lloyd, R.G.; Bolt, E.; Orren, D.K. Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 2007, 35, 5729–5747. [Google Scholar] [CrossRef] [Green Version]
- Ralf, C.; Hickson, I.D.; Wu, L. The Bloom′s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006, 281, 22839–22846. [Google Scholar] [CrossRef] [Green Version]
- Kopel, V.; Pozner, A.; Baran, N.; Manor, H. Unwinding of the third strand of a DNA triple helix, a novel activity of the SV40 large T-antigen helicase. Nucleic Acids Res. 1996, 24, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr.; Majumdar, A.; Desai, S.; Hickson, I.D.; Bohr, V.A.; Seidman, M.M. Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J. Biol. Chem. 2001, 276, 3024–3030. [Google Scholar] [CrossRef] [Green Version]
- Dixon, B.P.; Lu, L.; Chu, A.; Bissler, J.J. RecQ and RecG helicases have distinct roles in maintaining the stability of polypurine.polypyrimidine sequences. Mutat. Res. 2008, 643, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Bacolla, A.; Chakraborty, P.; Grosse, F.; Vasquez, K.M. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry 2010, 49, 6992–6999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Bacolla, A.; Del Mundo, I.M.; Zhao, J.; Wang, G.; Vasquez, K.M. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013, 41, 10345–10357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Hundseth, K.; Ding, H.; Vidhyasagar, V.; Inoue, A.; Nguyen, C.H.; Zain, R.; Lee, J.S.; Wu, Y. A distinct triplex DNA unwinding activity of ChlR1 helicase. J. Biol. Chem. 2015, 290, 5174–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishburn, J.; Tomko, E.; Galburt, E.; Hahn, S. Double-Stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3961–3966. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Maizels, N. Substrate-Specific inhibition of RecQ helicase. Nucleic Acids Res. 2001, 29, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom′s syndrome helicase unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Bennett, R.J.; Maizels, N. The Saccharomyces cerevisiae Sgs1 helicase efficiently unwinds G-G paired DNAs. Nucleic Acids Res. 1999, 27, 1978–1984. [Google Scholar] [CrossRef]
- Fry, M.; Loeb, L.A. Human Werner syndrome DNA helicase unwinds tetrahelical structures of the Fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem. 1999, 274, 12797–12802. [Google Scholar] [CrossRef] [Green Version]
- Bharti, S.K.; Sommers, J.A.; George, F.; Kuper, J.; Hamon, F.; Shin-ya, K.; Teulade-Fichou, M.P.; Kisker, C.; Brosh, R.M., Jr. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013, 288, 28217–28229. [Google Scholar] [CrossRef] [Green Version]
- Dehghani-Tafti, S.; Levdikov, V.; Antson, A.A.; Bax, B.; Sanders, C.M. Structural and functional analysis of the nucleotide and DNA binding activities of the human PIF1 helicase. Nucleic Acids Res. 2019, 47, 3208–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannier, J.B.; Sandhu, S.; Petalcorin, M.I.; Wu, X.; Nabi, Z.; Ding, H.; Boulton, S.J. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 2013, 342, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sommers, J.A.; Khan, I.; de Winter, J.P.; Brosh, R.M., Jr. Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 2012, 287, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, T.M.; Chao, K.; Green, J.M.; Sage, S.; Runyon, G.T. Large-scale purification and characterization of the Escherichia coli Rep gene product. J. Biol. Chem. 1989, 264, 10139–10147. [Google Scholar]
- Wong, I.; Lohman, T.M. Allosteric effects of nucleotide cofactors on Escherichia coli Rep helicase-DNA binding. Science 1992, 256, 350–355. [Google Scholar] [CrossRef]
- Chao, K.; Lohman, T.M. DNA and nucleotide-induced conformational changes in the Escherichia coli Rep and helicase II (UvrD) proteins. J. Biol. Chem. 1990, 265, 1067–1076. [Google Scholar]
- Lohman, T.M.; Bjornson, K.P. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 1996, 65, 169–214. [Google Scholar] [CrossRef]
- Wu, Y.; Sommers, J.A.; Loiland, J.A.; Kitao, H.; Kuper, J.; Kisker, C.; Brosh, R.M., Jr. The Q motif of Fanconi anemia group J protein (FANCJ) DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J. Biol. Chem. 2012, 287, 21699–21716. [Google Scholar] [CrossRef] [Green Version]
- Bird, L.E.; Brannigan, J.A.; Subramanya, H.S.; Wigley, D.B. Characterisation of Bacillus stearothermophilus PcrA helicase: Evidence against an active rolling mechanism. Nucleic Acids Res. 1998, 26, 2686–2693. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yang, W. UvrD helicase unwinds DNA one base pair at a time by a two-Part power stroke. Cell 2006, 127, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Sikora, B.; Eoff, R.L.; Matson, S.W.; Raney, K.D. DNA unwinding by Escherichia coli DNA helicase I (TraI) provides evidence for a processive monomeric molecular motor. J. Biol. Chem. 2006, 281, 36110–36116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tackett, A.J.; Chen, Y.; Cameron, C.E.; Raney, K.D. Multiple full-Length NS3 molecules are required for optimal unwinding of oligonucleotide DNA in vitro. J. Biol. Chem. 2005, 280, 10797–10806. [Google Scholar] [CrossRef] [Green Version]
- Eoff, R.L.; Raney, K.D. Kinetic mechanism for DNA unwinding by multiple molecules of Dda helicase aligned on DNA. Biochemistry 2010, 49, 4543–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, A.K.; Raney, K.D. Protein displacement by an assembly of helicase molecules aligned along single-Stranded DNA. Nat. Struct. Mol. Biol. 2004, 11, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Bedinger, P.; Hochstrasser, M.; Jongeneel, C.V.; Alberts, B.M. Properties of the T4 bacteriophage DNA replication apparatus: the T4 dda DNA helicase is required to pass a bound RNA polymerase molecule. Cell 1983, 34, 115–123. [Google Scholar] [CrossRef]
- Yancey-Wrona, J.E.; Matson, S.W. Bound Lac repressor protein differentially inhibits the unwinding reactions catalyzed by DNA helicases. Nucleic Acids Res. 1992, 20, 6713–6721. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.P.; Atkinson, J.; Gupta, M.K.; Mahdi, A.A.; Gwynn, E.J.; Rudolph, C.J.; Moon, P.B.; van Knippenberg, I.C.; Cadman, C.J.; Dillingham, M.S.; et al. Rep provides a second motor at the replisome to promote duplication of protein-Bound DNA. Mol. Cell 2009, 36, 654–666. [Google Scholar] [CrossRef]
- Bruning, J.G.; Howard, J.L.; McGlynn, P. Accessory replicative helicases and the replication of protein-Bound DNA. J. Mol. Biol. 2014, 426, 3917–3928. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr.; Li, J.L.; Kenny, M.K.; Karow, J.K.; Cooper, M.P.; Kureekattil, R.P.; Hickson, I.D.; Bohr, V.A. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem. 2000, 275, 23500–23508. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr.; Orren, D.K.; Nehlin, J.O.; Ravn, P.H.; Kenny, M.K.; Machwe, A.; Bohr, V.A. Functional and physical interaction between WRN helicase and human Replication protein A. J. Biol. Chem. 1999, 274, 18341–18350. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Sharma, S.; Sommers, J.A.; Kenny, M.K.; Cantor, S.B.; Brosh, R.M., Jr. FANCJ (BACH1) helicase forms DNA damage inducible foci with Replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood 2007, 110, 2390–2398. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.C.; Gray, M.D.; Oshima, J.; Loeb, L.A. Characterization of Werner syndrome protein DNA helicase activity: Directionality, substrate dependence and stimulation by Replication protein A. Nucleic Acids Res. 1998, 26, 2879–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, J.A.; Banerjee, T.; Hinds, T.; Wan, B.; Wold, M.S.; Lei, M.; Brosh, R.M., Jr. Novel function of the Fanconi anemia group J or RECQ1 helicase to disrupt protein-DNA complexes in a Replication protein A-Stimulated manner. J. Biol. Chem. 2014, 289, 19928–19941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Picha, K.M. Structure and function of hexameric helicases. Annu. Rev. Biochem. 2000, 69, 651–697. [Google Scholar] [CrossRef]
- Trakselis, M.A. Structural mechanisms of hexameric helicase loading, assembly, and unwinding. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.L.; Davey, M.J.; O’Donnell, M. Mcm4,6,7 uses a “pump in ring” mechanism to unwind DNA by steric exclusion and actively translocate along a duplex. J. Biol. Chem. 2003, 278, 49171–49182. [Google Scholar] [CrossRef] [Green Version]
- Bonhoeffer, F. DNA transfer and DNA synthesis during bacterial conjugation. Z. Vererb. 1966, 98, 141–149. [Google Scholar] [CrossRef]
- Carl, P.L. Escherichia coli mutants with temperature-sensitive synthesis of DNA. Mol. Gen. Genet. 1970, 109, 107–122. [Google Scholar] [CrossRef]
- Fangman, W.L.; Novick, A. Characterization of two bacterial mutants with temperature-Sensitive synthesis of DNA. Genetics 1968, 60, 1–17. [Google Scholar] [PubMed]
- Hirota, Y.; Ryter, A.; Jacob, F. Thermosensitive mutants of E. coli affected in the processes of DNA synthesis and cellular division. Cold Spring Harb. Symp. Quant. Biol. 1968, 33, 677–693. [Google Scholar] [CrossRef]
- Wechsler, J.A.; Gross, J.D. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol. Gen. Genet. 1971, 113, 273–284. [Google Scholar] [CrossRef]
- LeBowitz, J.H.; McMacken, R. The Escherichia coli DnaB replication protein is a DNA helicase. J. Biol. Chem. 1986, 261, 4738–4748. [Google Scholar]
- Baker, T.A.; Funnell, B.E.; Kornberg, A. Helicase action of dnaB protein during replication from the Escherichia coli chromosomal origin in vitro. J. Biol. Chem. 1987, 262, 6877–6885. [Google Scholar] [PubMed]
- Kornberg, A. Ten commandments: Lessons from the enzymology of DNA replication. J. Bacteriol. 2000, 182, 3613–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochman, M.L.; Schwacha, A. The Mcm complex: Unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. MMBR 2009, 73, 652–683. [Google Scholar] [CrossRef] [Green Version]
- Maine, G.T.; Sinha, P.; Tye, B.K. Mutants of S. cerevisiae defective in the maintenance of minichromosomes. Genetics 1984, 106, 365–385. [Google Scholar]
- Hennessy, K.M.; Lee, A.; Chen, E.; Botstein, D. A group of interacting yeast DNA replication genes. Genes Dev. 1991, 5, 958–969. [Google Scholar] [CrossRef] [Green Version]
- Schwacha, A.; Bell, S.P. Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol. Cell 2001, 8, 1093–1104. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamada, H.; Yanagida, M. Fission yeast minichromosome loss mutants mis cause lethal aneuploidy and replication abnormality. Mol. Biol. Cell 1994, 5, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Blow, J.J.; Laskey, R.A. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 1988, 332, 546–548. [Google Scholar] [CrossRef]
- Chong, J.P.; Blow, J.J. DNA replication licensing factor. Prog. Cell Cycle Res. 1996, 2, 83–90. [Google Scholar] [PubMed]
- Thommes, P.; Kubota, Y.; Takisawa, H.; Blow, J.J. The RLF-M component of the replication licensing system forms complexes containing all six MCM/P1 polypeptides. EMBO J. 1997, 16, 3312–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelman, L.M.; O’Dell, W.B.; Kelman, Z. Unwinding twenty years of the archaeal minichromosome maintenance helicase. J. Bacteriol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J. Biol. Chem. 1997, 272, 24508–24513. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011, 18, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Deegan, T.D.; Diffley, J.F. MCM: One ring to rule them all. Curr. Opin. Struct. Biol. 2016, 37, 145–151. [Google Scholar] [CrossRef]
- Zhai, Y.; Tye, B.K. Structure of the MCM2-7 double hexamer and its implications for the mechanistic functions of the Mcm2-7 complex. Adv. Exp. Med. Biol. 2017, 1042, 189–205. [Google Scholar] [CrossRef]
- Trakselis, M.A.; Seidman, M.M.; Brosh, R.M., Jr. Mechanistic insights into how CMG helicase facilitates replication past DNA roadblocks. DNA Repair 2017, 55, 76–82. [Google Scholar] [CrossRef]
- Meagher, M.; Epling, L.B.; Enemark, E.J. DNA translocation mechanism of the MCM complex and implications for replication initiation. Nat. Commun. 2019, 10, 3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangelo, I.A.; Hough, P.V.; Wall, J.S.; Dodson, M.; Dean, F.B.; Hurwitz, J. ATP-dependent assembly of double hexamers of SV40 T antigen at the viral origin of DNA replication. Nature 1989, 338, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Egelman, E.H.; Yu, X.; Wild, R.; Hingorani, M.M.; Patel, S.S. Bacteriophage T7 helicase/primase proteins form rings around single-stranded DNA that suggest a general structure for hexameric helicases. Proc. Natl. Acad. Sci. USA 1995, 92, 3869–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, S.; Eliason, W.K.; Steitz, T.A. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 2007, 318, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewster, A.S.; Wang, G.; Yu, X.; Greenleaf, W.B.; Carazo, J.M.; Tjajadi, M.; Klein, M.G.; Chen, X.S. Crystal structure of a near-full-length archaeal MCM: Functional insights for an AAA+ hexameric helicase. Proc. Natl. Acad. Sci. USA 2008, 105, 20191–20196. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhai, Y.; Zhang, Y.; Li, W.; Yang, M.; Lei, J.; Tye, B.K.; Gao, N. Structure of the eukaryotic MCM complex at 3.8 A. Nature 2015, 524, 186–191. [Google Scholar] [CrossRef]
- Li, H.; O’Donnell, M.E. The eukaryotic CMG helicase at the replication fork: Emerging architecture reveals an unexpected mechanism. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40. [Google Scholar] [CrossRef]
- Bernstein, D.A.; Zittel, M.C.; Keck, J.L. High-Resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003, 22, 4910–4921. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; Ababou, M.; Onclercq, R.; Dutertre, S.; Chadli, E.; Hda, N.; Benslimane, A.; Amor-Gueret, M. Identification of a novel BLM missense mutation (2706T>C) in a Moroccan patient with Bloom’s syndrome. Hum. Mutat. 2000, 15, 584–585. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Rong, S.B.; Valiaho, J.; Vihinen, M. Structural basis of Bloom syndrome (BS) causing mutations in the BLM helicase domain. Mol. Med. 2000, 6, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Gogol, E.P.; Seifried, S.E.; von Hippel, P.H. Structure and assembly of the Escherichia coli transcription termination factor rho and its interaction with RNA. I. Cryoelectron microscopic studies. J. Mol. Biol. 1991, 221, 1127–1138. [Google Scholar] [CrossRef]
- Stasiak, A.; Tsaneva, I.R.; West, S.C.; Benson, C.J.; Yu, X.; Egelman, E.H. The Escherichia coli RuvB branch migration protein forms double hexameric rings around DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 7618–7622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machius, M.; Henry, L.; Palnitkar, M.; Deisenhofer, J. Crystal structure of the DNA nucleotide excision repair enzyme UvrB from Thermus thermophilus. Proc. Natl. Acad. Sci. USA 1999, 96, 11717–11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, K.; Chen, P.J.; Skorvaga, M.; Van Houten, B.; Kisker, C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999, 18, 6899–6907. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Guo, S.; Tabor, S.; Richardson, C.C.; Ellenberger, T. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell 1999, 99, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Singleton, M.R.; Sawaya, M.R.; Ellenberger, T.; Wigley, D.B. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 2000, 101, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Singleton, M.R.; Dillingham, M.S.; Gaudier, M.; Kowalczykowski, S.C.; Wigley, D.B. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 2004, 432, 187–193. [Google Scholar] [CrossRef]
- Enemark, E.J.; Joshua-Tor, L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 2006, 442, 270–275. [Google Scholar] [CrossRef]
- Liu, H.; Rudolf, J.; Johnson, K.A.; McMahon, S.A.; Oke, M.; Carter, L.; McRobbie, A.M.; Brown, S.E.; Naismith, J.H.; White, M.F. Structure of the DNA repair helicase XPD. Cell 2008, 133, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Fuss, J.O.; Cheng, Q.J.; Arvai, A.S.; Hammel, M.; Roberts, V.A.; Cooper, P.K.; Tainer, J.A. XPD helicase structures and activities: Insights into the cancer and aging phenotypes from XPD mutations. Cell 2008, 133, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Wolski, S.C.; Kuper, J.; Hanzelmann, P.; Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008, 6, e149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, A.C.; Shrestha, B.; Popuri, V.; Burgess-Brown, N.; Muzzolini, L.; Costantini, S.; Vindigni, A.; Gileadi, O. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc. Natl. Acad. Sci. USA 2009, 106, 1039–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.A.; Savitsky, P.; Allerston, C.K.; Bizard, A.H.; Ozer, O.; Sarlos, K.; Liu, Y.; Pardon, E.; Steyaert, J.; Hickson, I.D.; et al. Crystal structure of the Bloom’s syndrome helicase indicates a role for the HRDC domain in conformational changes. Nucleic Acids Res. 2015, 43, 5221–5235. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.C.; Gomathinayagam, S.; Swuec, P.; Berti, M.; Zhang, Y.; Schnecke, C.; Marino, F.; von Delft, F.; Renault, L.; Costa, A.; et al. Human RECQ1 helicase-driven DNA unwinding, annealing, and branch migration: Insights from DNA complex structures. Proc. Natl. Acad. Sci. USA 2015, 112, 4286–4291. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Bai, L.; Sun, J.; Georgescu, R.; Liu, J.; O’Donnell, M.E.; Li, H. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat. Struct. Mol. Biol. 2016, 23, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, S.; Sauer, F.; Kisker, C. The structural and functional characterization of human RecQ4 reveals insights into its helicase mechanism. Nat. Commun. 2017, 8, 15907. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Aitkenhead, H.; Savitsky, P.; Gileadi, O. Insights into the RecQ helicase mechanism revealed by the structure of the helicase domain of human RECQL5. Nucleic Acids Res. 2017, 45, 4231–4243. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Wigley, D.B. DNA translocation mechanism of an XPD family helicase. eLife 2018, 7. [Google Scholar] [CrossRef]
- Lu, K.Y.; Chen, W.F.; Rety, S.; Liu, N.N.; Wu, W.Q.; Dai, Y.X.; Li, D.; Ma, H.Y.; Dou, S.X.; Xi, X.G. Insights into the structural and mechanistic basis of multifunctional S. cerevisiae Pif1p helicase. Nucleic Acids Res. 2018, 46, 1486–1500. [Google Scholar] [CrossRef] [Green Version]
- Voter, A.F.; Qiu, Y.; Tippana, R.; Myong, S.; Keck, J.L. A guanine-flipping and sequestration mechanism for G-Quadruplex unwinding by RecQ helicases. Nat. Commun. 2018, 9, 4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, N.; Byrd, A.K.; Bharath, S.R.; Yang, O.; Jia, Y.; Tang, X.; Ha, T.; Raney, K.D.; Song, H. Structural basis for DNA unwinding at forked dsDNA by two coordinating Pif1 helicases. Nat. Commun. 2019, 10, 5375. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.R.; Brewer, L.R.; Corzett, M.; Balhorn, R.; Yeh, Y.; Kowalczykowski, S.C.; Baskin, R.J. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature 2001, 409, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Dohoney, K.M.; Gelles, J. Chi-sequence recognition and DNA translocation by single RecBCD helicase/nuclease molecules. Nature 2001, 409, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Rasnik, I.; Cheng, W.; Babcock, H.P.; Gauss, G.H.; Lohman, T.M.; Chu, S. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 2002, 419, 638–641. [Google Scholar] [CrossRef]
- Mohapatra, S.; Lin, C.T.; Feng, X.A.; Basu, A.; Ha, T. Single-molecule analysis and engineering of DNA motors. Chem. Rev. 2019. [Google Scholar] [CrossRef]
- Sun, B. Rescuing replication from barriers: Mechanistic insights from single-molecule studies. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Wang, M.D. Single-Molecule perspectives on helicase mechanisms and functions. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 15–25. [Google Scholar] [CrossRef]
- Myong, S.; Rasnik, I.; Joo, C.; Lohman, T.M.; Ha, T. Repetitive shuttling of a motor protein on DNA. Nature 2005, 437, 1321–1325. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Michaelis, J. Mechanisms of nucleic acid translocases: Lessons from structural biology and single-Molecule biophysics. Curr. Opin. Struct. Biol. 2007, 17, 87–95. [Google Scholar] [CrossRef]
- Lionnet, T.; Dawid, A.; Bigot, S.; Barre, F.X.; Saleh, O.A.; Heslot, F.; Allemand, J.F.; Bensimon, D.; Croquette, V. DNA mechanics as a tool to probe helicase and translocase activity. Nucleic Acids Res. 2006, 34, 4232–4244. [Google Scholar] [CrossRef] [Green Version]
- Rasnik, I.; Myong, S.; Ha, T. Unraveling helicase mechanisms one molecule at a time. Nucleic Acids Res. 2006, 34, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.S.; Bai, L.; Smith, B.Y.; Patel, S.S.; Wang, M.D. Single-molecule studies reveal dynamics of DNA unwinding by the ring-Shaped T7 helicase. Cell 2007, 129, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Dallmann, H.G.; McHenry, C.S.; Marians, K.J. Coupling of a replicative polymerase and helicase: A tau-DnaB interaction mediates rapid replication fork movement. Cell 1996, 84, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Stano, N.M.; Jeong, Y.J.; Donmez, I.; Tummalapalli, P.; Levin, M.K.; Patel, S.S. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 2005, 435, 370–373. [Google Scholar] [CrossRef]
- Hamdan, S.M.; Johnson, D.E.; Tanner, N.A.; Lee, J.B.; Qimron, U.; Tabor, S.; van Oijen, A.M.; Richardson, C.C. Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 2007, 27, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Spies, M.; Amitani, I.; Baskin, R.J.; Kowalczykowski, S.C. RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell 2007, 131, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.; Park, J.; Pugh, R.A.; Ha, T.; Spies, M. Single-Molecule analysis reveals differential effect of ssDNA-Binding proteins on DNA translocation by XPD helicase. Mol. Cell 2009, 35, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Myong, S.; Niedziela-Majka, A.; Lee, K.S.; Yu, J.; Lohman, T.M.; Ha, T. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell 2010, 142, 544–555. [Google Scholar] [CrossRef] [Green Version]
- Yardimci, H.; Wang, X.; Loveland, A.B.; Tappin, I.; Rudner, D.Z.; Hurwitz, J.; van Oijen, A.M.; Walter, J.C. Bypass of a protein barrier by a replicative DNA helicase. Nature 2012, 492, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, I.J.; Greene, E.C. Molecular traffic jams on DNA. Annu. Rev. Biophys. 2013, 42, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, J.L.; Chistol, G.; Gao, A.O.; Raschle, M.; Larsen, N.B.; Mann, M.; Duxin, J.P.; Walter, J.C. The CMG helicase bypasses DNA-protein cross-Links to facilitate their repair. Cell 2019, 176, 167–181.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticau, S.; Friedman, L.J.; Ivica, N.A.; Gelles, J.; Bell, S.P. Single-Molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 2015, 161, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harami, G.M.; Seol, Y.; In, J.; Ferencziova, V.; Martina, M.; Gyimesi, M.; Sarlos, K.; Kovacs, Z.J.; Nagy, N.T.; Sun, Y.; et al. Shuttling along DNA and directed processing of D-Loops by RecQ helicase support quality control of homologous recombination. Proc. Natl. Acad. Sci. USA 2017, 114, E466–E475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniecki, K.; De Tullio, L.; Greene, E.C. A change of view: Homologous recombination at single-molecule resolution. Nat. Rev. Genet. 2018, 19, 191–207. [Google Scholar] [CrossRef]
- Singh, S.P.; Soranno, A.; Sparks, M.A.; Galletto, R. Branched unwinding mechanism of the Pif1 family of DNA helicases. Proc. Natl. Acad. Sci. USA 2019, 116, 24533–24541. [Google Scholar] [CrossRef]
- Sancar, A.; Hearst, J.E. Molecular matchmakers. Science 1993, 259, 1415–1420. [Google Scholar] [CrossRef]
- Bagchi, D.; Manosas, M.; Zhang, W.; Manthei, K.A.; Hodeib, S.; Ducos, B.; Keck, J.L.; Croquette, V. Single molecule kinetics uncover roles for E. coli RecQ DNA helicase domains and interaction with SSB. Nucleic Acids Res. 2018, 46, 8500–8515. [Google Scholar] [CrossRef] [Green Version]
- Shereda, R.D.; Bernstein, D.A.; Keck, J.L. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 2007, 282, 19247–19258. [Google Scholar] [CrossRef] [Green Version]
- Shereda, R.D.; Reiter, N.J.; Butcher, S.E.; Keck, J.L. Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB’s C terminus. J. Mol. Biol. 2009, 386, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Doherty, K.M.; Sommers, J.A.; Gray, M.D.; Lee, J.W.; von Kobbe, C.; Thoma, N.H.; Kureekattil, R.P.; Kenny, M.K.; Brosh, R.M., Jr. Physical and functional mapping of the Replication protein A interaction domain of the Werner and Bloom syndrome helicases. J. Biol. Chem. 2005, 280, 29494–29505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.C.; Lao, Y.; Kamath-Loeb, A.; Wold, M.S.; Loeb, L.A. The N-Terminal domain of the large subunit of human Replication protein A binds to Werner syndrome protein and stimulates helicase activity. Mech. Ageing Dev. 2003, 124, 921–930. [Google Scholar] [CrossRef]
- Opresko, P.L.; Otterlei, M.; Graakjaer, J.; Bruheim, P.; Dawut, L.; Kolvraa, S.; May, A.; Seidman, M.M.; Bohr, V.A. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell 2004, 14, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.A.; Richardson, C.C. Characterization of the helicase and primase activities of the 63-kDa component of the bacteriophage T7 gene 4 protein. J. Biol. Chem. 1989, 264, 13066–13073. [Google Scholar] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Dillingham, M.S. Superfamily I helicases as modular components of DNA-Processing machines. Biochem. Soc. Trans. 2011, 39, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Dillingham, M.S.; Kowalczykowski, S.C. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. MMBR 2008, 72, 642–671, Table of Contents. [Google Scholar] [CrossRef] [Green Version]
- Estep, K.N.; Brosh, R.M., Jr. RecQ and Fe-S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochem. Soc. Trans. 2018, 46, 77–95. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Richardson, C.C. Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 2011, 15, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Nossal, N.G. Protein-Protein interactions at a DNA replication fork: Bacteriophage T4 as a model. FASEB J. 1992, 6, 871–878. [Google Scholar] [CrossRef]
- Rezazadeh, S. RecQ helicases; at the crossroad of genome replication, repair, and recombination. Mol. Biol. Rep. 2012, 39, 4527–4543. [Google Scholar] [CrossRef]
- Sharma, S.; Doherty, K.M.; Brosh, R.M., Jr. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem. J. 2006, 398, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-Protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Peng, M.; Guillemette, S.; Quan, S.; Maniatis, S.; Wu, Y.; Venkatesh, A.; Shaffer, S.A.; Brosh, R.M., Jr.; Cantor, S.B. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012, 8, e1002786. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, R.; Muftuoglu, M.; Bohr, V.A. The role of WRN in DNA repair is affected by post-translational modifications. Mech. Ageing Dev. 2007, 128, 50–57. [Google Scholar] [CrossRef]
- Bohm, S.; Bernstein, K.A. The role of post-Translational modifications in fine-tuning BLM helicase function during DNA repair. DNA Repair 2014, 22, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, J.A.; Suhasini, A.N.; Brosh, R.M., Jr. Protein degradation pathways regulate the functions of helicases in the DNA damage response and maintenance of genomic stability. Biomolecules 2015, 5, 590–616. [Google Scholar] [CrossRef] [Green Version]
- Bain, F.E.; Wu, C.G.; Spies, M. Single-molecule sorting of DNA helicases. Methods 2016, 108, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Masuda-Ozawa, T.; Hoang, T.; Seo, Y.S.; Chen, L.F.; Spies, M. Single-molecule sorting reveals how ubiquitylation affects substrate recognition and activities of FBH1 helicase. Nucleic Acids Res. 2013, 41, 3576–3587. [Google Scholar] [CrossRef]
- Kuper, J.; Kisker, C. DNA helicases in NER, BER, and MMR. Adv. Exp. Med. Biol. 2013, 767, 203–224. [Google Scholar] [CrossRef]
- Nakayama, H.; Nakayama, K.; Nakayama, R.; Irino, N.; Nakayama, Y.; Hanawalt, P.C. Isolation and genetic characterization of a thymineless death-Resistant mutant of Escherichia coli K12: Identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol. Gen. Genet. 1984, 195, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Khodursky, A.; Guzman, E.C.; Hanawalt, P.C. Thymineless death lives on: New insights into a classic phenomenon. Annu. Rev. Microbiol. 2015, 69, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Hanawalt, P.C. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 1999, 262, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, S.; McDonald, J.P.; Bendixen, C.; Arthur, L.; Rothstein, R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol. Cell. Biol. 1994, 14, 8391–8398. [Google Scholar] [CrossRef] [Green Version]
- Watt, P.M.; Louis, E.J.; Borts, R.H.; Hickson, I.D. Sgs1: A eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell 1995, 81, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; Mills, K.; Guarente, L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science 1997, 277, 1313–1316. [Google Scholar] [CrossRef]
- Stewart, E.; Chapman, C.R.; Al-Khodairy, F.; Carr, A.M.; Enoch, T. rqh1+, a fission yeast gene related to the Bloom’s and Werner’s syndrome genes, is required for reversible S phase arrest. EMBO J. 1997, 16, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.L.; Dixon, J.; Osman, F.; Whitby, M.C. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 2000, 19, 2751–2762. [Google Scholar] [CrossRef] [Green Version]
- Kaliraman, V.; Mullen, J.R.; Fricke, W.M.; Bastin-Shanower, S.A.; Brill, S.J. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001, 15, 2730–2740. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Suhasini, A.N.; Rawtani, N.A.; Wu, Y.; Sommers, J.A.; Sharma, S.; Mosedale, G.; North, P.S.; Cantor, S.B.; Hickson, I.D.; Brosh, R.M., Jr. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom′s syndrome. EMBO J. 2011, 30, 692–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.L.; North, P.S.; Dart, A.; Lakin, N.D.; Hickson, I.D. Phosphorylation of the Bloom′s syndrome helicase and its role in recovery from S-Phase arrest. Mol. Cell. Biol. 2004, 24, 1279–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, J.A.; Schleker, T.; Rojas, V.; Bjergbaek, L.; Tercero, J.A.; Gasser, S.M. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005, 19, 3055–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, S.L.; Shoemaker, D.D.; Boeke, J.D. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 2003, 35, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Kuo, H.K.; Savukoski, D.; Brodsky, M.H.; Sekelsky, J. Three structure-selective endonucleases are essential in the absence of BLM helicase in Drosophila. PLoS Genet. 2011, 7, e1002315. [Google Scholar] [CrossRef] [Green Version]
- Mullen, J.R.; Kaliraman, V.; Ibrahim, S.S.; Brill, S.J. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 2001, 157, 103–118. [Google Scholar]
- Stundon, J.L.; Zakian, V.A. Identification of Saccharomyces cerevisiae genes whose deletion causes synthetic effects in cells with reduced levels of the nuclear Pif1 DNA helicase. G3 2015, 5, 2913–2918. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, K.; McKim, K.; Brill, S.J.; Sekelsky, J. Synthetic lethality of Drosophila in the absence of the MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics 2007, 176, 1993–2001. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019. [Google Scholar] [CrossRef] [Green Version]
- Kategaya, L.; Perumal, S.K.; Hager, J.H.; Belmont, L.D. Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience 2019, 13, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieb, S.; Blaha-Ostermann, S.; Kamper, E.; Rippka, J.; Schwarz, C.; Ehrenhofer-Wolfer, K.; Schlattl, A.; Wernitznig, A.; Lipp, J.J.; Nagasaka, K.; et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-High tumor cells. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-Dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichierri, P.; Franchitto, A.; Mosesso, P.; Palitti, F. Werner′s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-Phase of cell cycle. Mol. Biol. Cell 2001, 12, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, K.K.; Sidorova, J.; Saintigny, Y.; Poot, M.; Gollahon, K.; Rabinovitch, P.S.; Monnat, R.J., Jr. Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell 2007, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008, 180, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidorova, J.M.; Li, N.; Folch, A.; Monnat, R.J., Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 2008, 7, 796–807. [Google Scholar] [CrossRef]
- Cheng, W.H.; Muftic, D.; Muftuoglu, M.; Dawut, L.; Morris, C.; Helleday, T.; Shiloh, Y.; Bohr, V.A. WRN is required for ATM activation and the S-Phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol. Biol. Cell 2008, 19, 3923–3933. [Google Scholar] [CrossRef] [Green Version]
- Basile, G.; Leuzzi, G.; Pichierri, P.; Franchitto, A. Checkpoint-Dependent and independent roles of the Werner syndrome protein in preserving genome integrity in response to mild replication stress. Nucleic Acids Res. 2014, 42, 12628–12639. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Sommers, J.A.; Huang, J.; Seidman, M.M.; Brosh, R.M., Jr. Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. 2015, 25, 2830–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, S.; Sharma, S. The DNA repair helicase RECQ1 has a checkpoint-dependent role in mediating DNA damage responses induced by gemcitabine. J. Biol. Chem. 2019, 294, 15330–15345. [Google Scholar] [CrossRef] [PubMed]
- Urban, V.; Dobrovolna, J.; Huhn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Huhn, D.; et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 2017, 66, 658.e658–671.e658. [Google Scholar] [CrossRef] [Green Version]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-Transcriptional R-loops. Mol. Cell 2019. [Google Scholar] [CrossRef]
- Gong, Z.; Kim, J.E.; Leung, C.C.; Glover, J.N.; Chen, J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol. Cell 2010, 37, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Barthelemy, J.; Hanenberg, H.; Leffak, M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016, 44, 6803–6816. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef] [Green Version]
- Bharti, S.K.; Sommers, J.A.; Awate, S.; Bellani, M.A.; Khan, I.; Bradley, L.; King, G.A.; Seol, Y.; Vidhyasagar, V.; Wu, Y.; et al. A minimal threshold of FANCJ helicase activity is required for its response to replication stress or double-Strand break repair. Nucleic Acids Res. 2018, 46, 6238–6256. [Google Scholar] [CrossRef] [Green Version]
- Bansbach, C.E.; Betous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Bredemeyer, A.L.; Sowa, M.E.; Terret, M.E.; Jallepalli, P.V.; Harper, J.W.; Elledge, S.J. The SIOD disorder protein SMARCAL1 is an RPA-Interacting protein involved in replication fork restart. Genes Dev. 2009, 23, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, L.A.; Cortez, D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Coin, F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair 2011, 10, 714–721. [Google Scholar] [CrossRef]
- Compe, E.; Egly, J.M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef]
- Kuper, J.; Braun, C.; Elias, A.; Michels, G.; Sauer, F.; Schmitt, D.R.; Poterszman, A.; Egly, J.M.; Kisker, C. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 2014, 12, e1001954. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Izumikawa, K.; Yanagida, M.; Hayano, T.; Tachikawa, H.; Komatsu, W.; Shimamoto, A.; Futami, K.; Furuichi, Y.; Shinkawa, T.; Yamauchi, Y.; et al. Association of human DNA helicase RecQ5beta with RNA polymerase II and its possible role in transcription. Biochem. J. 2008, 413, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xu, X.; Liu, Y. The SET2-RPB1 interaction domain of human RECQ5 is important for transcription-associated genome stability. Mol. Cell. Biol. 2011, 31, 2090–2099. [Google Scholar] [CrossRef] [Green Version]
- Aygun, O.; Xu, X.; Liu, Y.; Takahashi, H.; Kong, S.E.; Conaway, R.C.; Conaway, J.W.; Svejstrup, J.Q. Direct inhibition of RNA polymerase II transcription by RECQL5. J. Biol. Chem. 2009, 284, 23197–23203. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Fox, D., 3rd; Guo, R.; Enomoto, T.; Wang, W. RecQL5 promotes genome stabilization through two parallel mechanisms--Interacting with RNA polymerase II and acting as a helicase. Mol. Cell. Biol. 2010, 30, 2460–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagaraj, R.; Huehn, D.; MacKellar, A.; Menigatti, M.; Zheng, L.; Urban, V.; Shevelev, I.; Greenleaf, A.L.; Janscak, P. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010, 38, 8131–8140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassube, S.A.; Jinek, M.; Fang, J.; Tsutakawa, S.; Nogales, E. Structural mimicry in transcription regulation of human RNA polymerase II by the DNA helicase RECQL5. Nat. Struct. Mol. Biol. 2013, 20, 892–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saponaro, M.; Kantidakis, T.; Mitter, R.; Kelly, G.P.; Heron, M.; Williams, H.; Soding, J.; Stewart, A.; Svejstrup, J.Q. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014, 157, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Pokharel, S.; Wang, J.T.; Xu, X.; Liu, Y. RECQ5-dependent SUMOylation of DNA topoisomerase I prevents transcription-associated genome instability. Nat. Commun. 2015, 6, 6720. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Cao, K.; Ryvkin, P.; Wang, L.S.; Johnson, F.B. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-Quadruplex forming potential. Nucleic Acids Res. 2010, 38, 1114–1122. [Google Scholar] [CrossRef]
- Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Nguyen, G.H.; Oshima, J.; Maizels, N.; Harris, C.C.; Monnat, R.J., Jr. The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum. Mol. Genet. 2016, 25, 2060–2069. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.H.; Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Welsh, J.A.; Schetter, A.J.; Kumamoto, K.; Wang, X.W.; Hickson, I.D.; et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-Quadruplex DNA motifs. Proc. Natl. Acad. Sci. USA 2014, 111, 9905–9910. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Parvathaneni, S.; Li, X.L.; Lal, A.; Sharma, S. Transcriptome guided identification of novel functions of RECQ1 helicase. Methods 2016, 108, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Ligabue, A.; Rogers, K.J.; Akey, J.M.; Monnat, R.J., Jr. Human RECQ helicase pathogenic variants, population variation and “missing” diseases. Hum. Mutat. 2017, 38, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Lyon, G.J.; Wang, K. Identifying disease mutations in genomic medicine settings: Current challenges and how to accelerate progress. Genome Med. 2012, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.A.; Salazar, E.P.; Stewart, S.A.; Thompson, L.H. ERCC2: cDNA cloning and molecular characterization of a human nucleotide excision repair gene with high homology to yeast RAD3. EMBO J. 1990, 9, 1437–1447. [Google Scholar] [CrossRef]
- Lehmann, A.R. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases. Genes Dev. 2001, 15, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.; Bailly, V.; Weber, C.; Thompson, L.H.; Prakash, L.; Prakash, S. Human xeroderma pigmentosum group D gene encodes a DNA helicase. Nature 1993, 365, 852–855. [Google Scholar] [CrossRef]
- Mathieu, N.; Kaczmarek, N.; Ruthemann, P.; Luch, A.; Naegeli, H. DNA quality control by a lesion sensor pocket of the Xeroderma pigmentosum group D helicase subunit of TFIIH. Curr. Biol. CB 2013, 23, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugasawa, K.; Akagi, J.; Nishi, R.; Iwai, S.; Hanaoka, F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, S.; Coin, F. Orchestral maneuvers at the damaged sites in nucleotide excision repair. Cell. Mol. Life Sci. CMLS 2015, 72, 2177–2186. [Google Scholar] [CrossRef]
- Houten, B.V.; Kuper, J.; Kisker, C. Role of XPD in cellular functions: To TFIIH and beyond. DNA Repair 2016, 44, 136–142. [Google Scholar] [CrossRef]
- Mui, T.P.; Fuss, J.O.; Ishida, J.P.; Tainer, J.A.; Barton, J.K. ATP-stimulated, DNA-Mediated redox signaling by XPD, a DNA repair and transcription helicase. J. Am. Chem. Soc. 2011, 133, 16378–16381. [Google Scholar] [CrossRef] [Green Version]
- Sontz, P.A.; Mui, T.P.; Fuss, J.O.; Tainer, J.A.; Barton, J.K. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1856–1861. [Google Scholar] [CrossRef] [Green Version]
- Weeda, G.; van Ham, R.C.; Masurel, R.; Westerveld, A.; Odijk, H.; de Wit, J.; Bootsma, D.; van der Eb, A.J.; Hoeijmakers, J.H. Molecular cloning and biological characterization of the human excision repair gene ERCC-3. Mol. Cell. Biol. 1990, 10, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeda, G.; van Ham, R.C.; Vermeulen, W.; Bootsma, D.; van der Eb, A.J.; Hoeijmakers, J.H. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders Xeroderma pigmentosum and Cockayne’s syndrome. Cell 1990, 62, 777–791. [Google Scholar] [CrossRef] [Green Version]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on Xeroderma pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom′s syndrome: Clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Bachrati, C.Z.; Borts, R.H.; Hickson, I.D. Mobile D-Loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006, 34, 2269–2279. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Nimonkar, A.V.; Ozsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human Exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.B.; Hickson, I.D. RecQ helicases: Conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 2013, 767, 161–184. [Google Scholar] [CrossRef]
- Chan, K.L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [Green Version]
- German, J.; Sanz, M.M.; Ciocci, S.; Ye, T.Z.; Ellis, N.A. Syndrome-Causing mutations of the BLM gene in persons in the Bloom′s Syndrome Registry. Hum. Mutat. 2007, 28, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner′s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Li, B.; Gray, M.D.; Oshima, J.; Mian, I.S.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3’-->5’ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Monnat, R.J., Jr. Werner syndrome (WRN) gene variants and their association with altered function and age-Associated diseases. Ageing Res. Rev. 2018, 41, 82–97. [Google Scholar] [CrossRef]
- Monnat, R.J., Jr. Human RECQ helicases: Roles in DNA metabolism, mutagenesis and cancer biology. Semin. Cancer Biol. 2010, 20, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar] [CrossRef]
- Du, X.; Shen, J.; Kugan, N.; Furth, E.E.; Lombard, D.B.; Cheung, C.; Pak, S.; Luo, G.; Pignolo, R.J.; DePinho, R.A.; et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol. Cell. Biol. 2004, 24, 8437–8446. [Google Scholar] [CrossRef] [Green Version]
- Laud, P.R.; Multani, A.S.; Bailey, S.M.; Wu, L.; Ma, J.; Kingsley, C.; Lebel, M.; Pathak, S.; DePinho, R.A.; Chang, S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005, 19, 2560–2570. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 2004, 306, 1951–1953. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84. [Google Scholar] [CrossRef]
- Van Maldergem, L.; Siitonen, H.A.; Jalkh, N.; Chouery, E.; De Roy, M.; Delague, V.; Muenke, M.; Jabs, E.W.; Cai, J.; Wang, L.L.; et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 2006, 43, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siitonen, H.A.; Kopra, O.; Kaariainen, H.; Haravuori, H.; Winter, R.M.; Saamanen, A.M.; Peltonen, L.; Kestila, M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 2003, 12, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macris, M.A.; Krejci, L.; Bussen, W.; Shimamoto, A.; Sung, P. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair 2006, 5, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009, 28, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.L.; Ghosh, A.K.; Kulikowicz, T.; Croteau, D.L.; Bohr, V.A. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair 2010, 9, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Tandazo, W.; Smeets, M.F.; Murphy, V.; Liu, R.; Hodson, C.; Heierhorst, J.; Deans, A.J.; Walkley, C.R. ATP-dependent helicase activity is dispensable for the physiological functions of Recql4. PLoS Genet. 2019, 15, e1008266. [Google Scholar] [CrossRef]
- Smeets, M.F.; DeLuca, E.; Wall, M.; Quach, J.M.; Chalk, A.M.; Deans, A.J.; Heierhorst, J.; Purton, L.E.; Izon, D.J.; Walkley, C.R. The Rothmund-Thomson syndrome helicase RECQL4 is essential for hematopoiesis. J. Clin. Investig. 2014, 124, 3551–3565. [Google Scholar] [CrossRef] [Green Version]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef]
- Levran, O.; Attwooll, C.; Henry, R.T.; Milton, K.L.; Neveling, K.; Rio, P.; Batish, S.D.; Kalb, R.; Velleuer, E.; Barral, S.; et al. The BRCA1-Interacting helicase BRIP1 is deficient in Fanconi anemia. Nat. Genet. 2005, 37, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.; White, M.F. The DNA repair helicases XPD and FancJ have essential iron-Sulfur domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sommers, J.A.; Suhasini, A.N.; Leonard, T.; Deakyne, J.S.; Mazin, A.V.; Shin-Ya, K.; Kitao, H.; Brosh, R.M., Jr. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood 2010, 116, 3780–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosh, R.M., Jr.; Cantor, S.B. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi anemia. Front. Genet. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Voulgaridou, G.P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 2011, 711, 13–27. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Bellani, M.; Liu, Y.; Seidman, M.M. Fanconi Anemia: A DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res. Rev. 2017, 33, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.R.; Bakker, S.T.; Agarwal, S.; Jansen, M.; Grassman, E.; Godthelp, B.C.; Ali, A.M.; Du, C.H.; Rooimans, M.A.; Fan, Q.; et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood 2009, 114, 174–180. [Google Scholar] [CrossRef]
- Positional cloning of the Fanconi anaemia group A gene. Nat. Genet. 1996, 14, 324–328. [CrossRef] [PubMed]
- Basbous, J.; Constantinou, A. A tumor suppressive DNA translocase named FANCM. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Decaillet, C.; Delannoy, M.; Wu, L.; Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA 2008, 105, 16107–16112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gari, K.; Decaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef]
- Xue, Y.; Li, Y.; Guo, R.; Ling, C.; Wang, W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Benet. 2008, 17, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Delannoy, M.; Ling, C.; Daee, D.; Osman, F.; Muniandy, P.A.; Shen, X.; Oostra, A.B.; Du, H.; Steltenpool, J.; et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell 2010, 37, 865–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef]
- Huang, J.; Liu, S.; Bellani, M.A.; Thazhathveetil, A.K.; Ling, C.; de Winter, J.P.; Wang, Y.; Wang, W.; Seidman, M.M. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 2013, 52, 434–446. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M., Jr. Holding all the cards-How Fanconi Anemia proteins deal with replication stress and preserve genomic stability. Genes 2019, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Duxin, J.P.; Walter, J.C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 2015, 37, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. Off. J. Jpn. Environ. Mutagen Soc. 2016, 38, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swuec, P.; Costa, A. DNA replication and inter-Strand crosslink repair: Symmetric activation of dimeric nanomachines? Biophys. Chem. 2017, 225, 15–19. [Google Scholar] [CrossRef] [PubMed]
- van der Lelij, P.; Chrzanowska, K.H.; Godthelp, B.C.; Rooimans, M.A.; Oostra, A.B.; Stumm, M.; Zdzienicka, M.Z.; Joenje, H.; de Winter, J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010, 86, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballew, B.J.; Yeager, M.; Jacobs, K.; Giri, N.; Boland, J.; Burdett, L.; Alter, B.P.; Savage, S.A. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum. Genet. 2013, 132, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guen, T.; Jullien, L.; Touzot, F.; Schertzer, M.; Gaillard, L.; Perderiset, M.; Carpentier, W.; Nitschke, P.; Picard, C.; Couillault, G.; et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum. Mol. Genet. 2013, 22, 3239–3249. [Google Scholar] [CrossRef] [Green Version]
- Walne, A.J.; Vulliamy, T.; Kirwan, M.; Plagnol, V.; Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 2013, 92, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Vannier, J.B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.; Ding, H.; Boulton, S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Barber, L.J.; Youds, J.L.; Ward, J.D.; McIlwraith, M.J.; O′Neil, N.J.; Petalcorin, M.I.; Martin, J.S.; Collis, S.J.; Cantor, S.B.; Auclair, M.; et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 2008, 135, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Sarek, G.; Kotsantis, P.; Ruis, P.; Van Ly, D.; Margalef, P.; Borel, V.; Zheng, X.F.; Flynn, H.R.; Snijders, A.P.; Chowdhury, D.; et al. CDK phosphorylation of TRF2 controls t-Loop dynamics during the cell cycle. Nature 2019. [Google Scholar] [CrossRef]
- Alkhunaizi, E.; Brosh, R.M., Jr.; Alkuraya, F.S.; Chitayat, D. Warsaw Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Abe, T.; Ooka, M.; Kawasumi, R.; Miyata, K.; Takata, M.; Hirota, K.; Branzei, D. Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc. Natl. Acad. Sci. USA 2018, 115, 8412–8417. [Google Scholar] [CrossRef] [Green Version]
- Cali, F.; Bharti, S.K.; Di Perna, R.; Brosh, R.M., Jr.; Pisani, F.M. Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway. Nucleic Acids Res. 2016, 44, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Cortone, G.; Zheng, G.; Pensieri, P.; Chiappetta, V.; Tate, R.; Malacaria, E.; Pichierri, P.; Yu, H.; Pisani, F.M. Interaction of the Warsaw breakage syndrome DNA helicase DDX11 with the replication fork-protection factor Timeless promotes sister chromatid cohesion. PLoS Genet. 2018, 14, e1007622. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Grossi, E.; Marin-Bejar, O.; Bharti, S.K.; Raimondi, I.; Gonzalez, J.; Martinez-Herrera, D.J.; Athie, A.; Amadoz, A.; Brosh, R.M., Jr.; et al. A long noncoding RNA regulates sister chromatid cohesion. Mol. Cell 2016, 63, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Bloom, K. Liberating cohesin from cohesion. Genes Dev. 2017, 31, 2113–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skibbens, R.V. Condensins and cohesins-One of these things is not like the other! J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Hernandez, S.; Bermejo, R. Replisome-Cohesin interfacing: A molecular rerspective. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1800109. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Kang, T.H.; Reardon, J.T.; Lee, J.H.; Ozturk, N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010, 584, 2618–2625. [Google Scholar] [CrossRef] [Green Version]
- Unsal-Kacmaz, K.; Mullen, T.E.; Kaufmann, W.K.; Sancar, A. Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 2005, 25, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Wanrooij, S.; Falkenberg, M. The human mitochondrial replication fork in health and disease. Biochim. Biophys. Acta 2010, 1797, 1378–1388. [Google Scholar] [CrossRef] [Green Version]
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016, 38, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, M.J.; Humble, M.M.; Sharief, F.S.; Copeland, W.C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J. Biol. Chem. 2010, 285, 29690–29702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, B.; Farge, G.; Pardo-Hernandez, C.; Tangefjord, S.; Falkenberg, M. Structural basis for adPEO-Causing mutations in the mitochondrial TWINKLE helicase. Hum. Mol. Genet. 2019, 28, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-Like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebarth, T.D.; Farr, C.L.; Kaguni, L.S. Modular architecture of the hexameric human mitochondrial DNA helicase. J. Mol. Biol. 2007, 367, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Ziebarth, T.D.; Gonzalez-Soltero, R.; Makowska-Grzyska, M.M.; Nunez-Ramirez, R.; Carazo, J.M.; Kaguni, L.S. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J. Biol. Chem. 2010, 285, 14639–14647. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef]
- Moyer, C.L.; Ivanovich, J.; Gillespie, J.L.; Doberstein, R.; Radke, M.R.; Richardson, M.E.; Kaufmann, S.H.; Swisher, E.M.; Goodfellow, P.J. Rare BRIP1 missense alleles confer risk for ovarian and breast cancer. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-Penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-Deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib monotherapy in patients with deleterious germline BRCA-Mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musella, A.; Bardhi, E.; Marchetti, C.; Vertechy, L.; Santangelo, G.; Sassu, C.; Tomao, F.; Rech, F.; D’Amelio, R.; Monti, M.; et al. Rucaparib: An emerging PARP inhibitor for treatment of recurrent ovarian cancer. Cancer Treat. Rev. 2018, 66, 7–14. [Google Scholar] [CrossRef]
- Hengel, S.R.; Spies, M.A.; Spies, M. Small-Molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering new medicines targeting helicases: Challenges and recent progress. J. Biomol. Screen. 2013, 18, 761–781. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Iannascoli, C.; Pichierri, P.; Shoemaker, R.H.; Brosh, R.M., Jr. Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional Fanconi Anemia pathway. Cancer Res. 2013, 73, 5497–5507. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Hsieh, H.J.; Wang, L.; et al. Inhibition of DNA2 nuclease as a therapeutic strategy targeting replication stress in cancer cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A selective small molecule DNA2 inhibitor for sensitization of human cancer cells to chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maga, G.; Falchi, F.; Garbelli, A.; Belfiore, A.; Witvrouw, M.; Manetti, F.; Botta, M. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid-Glutamic acid-Alanine-Aspartic acid box polypeptide 3. J. Med. Chem. 2008, 51, 6635–6638. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Falchi, F.; Garbelli, A.; Samuele, A.; Bernardo, V.; Paolucci, S.; Baldanti, F.; Schenone, S.; Manetti, F.; Maga, G.; et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: Towards the next generation HIV-1 inhibitors. Bioorganic Med. Chem. Lett. 2012, 22, 2094–2098. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y.; Sugiyama, T.; Nakaya, R.; Kanamori, M.; Kohno, T.; Enomoto, T.; Chino, M. Effect of heliquinomycin on the activity of human minichromosome maintenance 4/6/7 helicase. FEBS J. 2009, 276, 3382–3391. [Google Scholar] [CrossRef]
- Simon, N.; Bochman, M.L.; Seguin, S.; Brodsky, J.L.; Seibel, W.L.; Schwacha, A. Ciprofloxacin is an inhibitor of the Mcm2-7 replicative helicase. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-Small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M., Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef]
Figure 1.
Timeline for discovery of prokaryotic, eukaryotic, and viral DNA helicases. See references and text for details. 1953, Discovery of DNA double helix [16]; 1958, Discovery of DNA polymerase I [17,18]; 1976, Discovery of first bacterial DNA unwinding enzyme [1,2,3]; 1978, “Helicase” term coined [6,7]; 1978, First eukaryotic DNA helicase [19]; 1982, First bacteriophage helicase [20]; 1985, First mammalian DNA helicase [21]; 1986, First mammalian viral DNA helicase [22]; 1986, First yeast DNA helicase [23]; 1990, First human DNA helicase [11]; 1992, First mitochondrial DNA helicase [12]; 1996, First chloroplast DNA helicase [13]; 2002, First malaria DNA helicase [24]; 2011, Genome-wide prediction of human DNA and RNA helicases [11].
Figure 1.
Timeline for discovery of prokaryotic, eukaryotic, and viral DNA helicases. See references and text for details. 1953, Discovery of DNA double helix [16]; 1958, Discovery of DNA polymerase I [17,18]; 1976, Discovery of first bacterial DNA unwinding enzyme [1,2,3]; 1978, “Helicase” term coined [6,7]; 1978, First eukaryotic DNA helicase [19]; 1982, First bacteriophage helicase [20]; 1985, First mammalian DNA helicase [21]; 1986, First mammalian viral DNA helicase [22]; 1986, First yeast DNA helicase [23]; 1990, First human DNA helicase [11]; 1992, First mitochondrial DNA helicase [12]; 1996, First chloroplast DNA helicase [13]; 2002, First malaria DNA helicase [24]; 2011, Genome-wide prediction of human DNA and RNA helicases [11].

Figure 2.
Representative assays to measure helicase-catalyzed DNA unwinding. Dates of published papers using the depicted techniques are indicated in blue font. (A) S1 nuclease digestion assay to measure DNA helicase activity. In the example shown, products from helicase reactions containing duplex [32P] DNA are incubated with single-strand-specific S1 nuclease and precipitated with cold trichloroacetic acid on membrane filter discs [1]. Total acid-precipitated [32P] label was determined by scintillation counting to indirectly measure DNA unwinding. Note: The cartoon schematic depicts small, acid-soluble pieces of ssDNA. (B) Native polyacrylamide gel resolution of helicase reaction products. In the example shown, an M13 partial duplex substrate with an annealed [32P] DNA fragment (115-mer) was incubated with dTTP and purified bacteriophage T7 gene 4 helicase protein [42]. The helicase reaction products were resolved on a nondenaturing 8% polyacrylamide gel, followed by autoradiography of the wet gel. Scintillation counting of excised radioactive gel slices corresponding to the DNA substrate and unwound ssDNA product would be performed to quantitate helicase activity. A similar strand displacement assay to measure helicase activity in vitro was developed by Nancy Nossal’s laboratory [20] (see text). Interestingly, during the authors’ time together in the Matson laboratory when Brosh was doing research for his PhD thesis at the University of North Carolina at Chapel Hill (1991–1996), PhosphorImager technology largely replaced film autoradiography and scintillation counting for visualization and quantitative analysis of helicase activity measured in vitro. (C) Fluorescence resonance energy transfer to monitor helicase-catalyzed DNA unwinding continuously. In the example shown, upon separation of the two complementary strands fluorescence emission from fluorescein (F) excitation can be monitored by a photosensor because it is not quenched by the hexachlorofluorescein (HF) [43]. Data can be collected in real-time by fluorescence instrumentation equipped with a stopped-flow device. (D) Single-molecule detection of helicase-catalyzed DNA unwinding using optical tweezers. In the example shown, DNA unwinding is detected by the movement of the magnetic bead subjected to a force (F) induced by a magnetic field gradient [44]. Helicase-catalyzed DNA unwinding results in a stretched single-strand (molecule extension (L)) which enables the magnetic bead to migrate toward the external magnet away from the immobilized DNA end tethered to the coverslip. Video-tracking is employed to detect and measure migration of the microsphere. (E) Combined dual-trap optical tweezers and SM fluorescence microscopy to simultaneously detect and measure unwinding activity and conformation of a DNA helicase protein site-specifically labeled with a fluorophore(s) [45]. The figure depicts an experimental system with the two indicated microspheres in dual optical traps tethered by a DNA hairpin. The confocal microscope (green) detects the conformation of the fluorescently labeled helicase protein complex actively unwinding the duplex DNA hairpin. Reference [46] provides a detailed protocol describing this technique dubbed high-resolution “fleezers”.
Figure 2.
Representative assays to measure helicase-catalyzed DNA unwinding. Dates of published papers using the depicted techniques are indicated in blue font. (A) S1 nuclease digestion assay to measure DNA helicase activity. In the example shown, products from helicase reactions containing duplex [32P] DNA are incubated with single-strand-specific S1 nuclease and precipitated with cold trichloroacetic acid on membrane filter discs [1]. Total acid-precipitated [32P] label was determined by scintillation counting to indirectly measure DNA unwinding. Note: The cartoon schematic depicts small, acid-soluble pieces of ssDNA. (B) Native polyacrylamide gel resolution of helicase reaction products. In the example shown, an M13 partial duplex substrate with an annealed [32P] DNA fragment (115-mer) was incubated with dTTP and purified bacteriophage T7 gene 4 helicase protein [42]. The helicase reaction products were resolved on a nondenaturing 8% polyacrylamide gel, followed by autoradiography of the wet gel. Scintillation counting of excised radioactive gel slices corresponding to the DNA substrate and unwound ssDNA product would be performed to quantitate helicase activity. A similar strand displacement assay to measure helicase activity in vitro was developed by Nancy Nossal’s laboratory [20] (see text). Interestingly, during the authors’ time together in the Matson laboratory when Brosh was doing research for his PhD thesis at the University of North Carolina at Chapel Hill (1991–1996), PhosphorImager technology largely replaced film autoradiography and scintillation counting for visualization and quantitative analysis of helicase activity measured in vitro. (C) Fluorescence resonance energy transfer to monitor helicase-catalyzed DNA unwinding continuously. In the example shown, upon separation of the two complementary strands fluorescence emission from fluorescein (F) excitation can be monitored by a photosensor because it is not quenched by the hexachlorofluorescein (HF) [43]. Data can be collected in real-time by fluorescence instrumentation equipped with a stopped-flow device. (D) Single-molecule detection of helicase-catalyzed DNA unwinding using optical tweezers. In the example shown, DNA unwinding is detected by the movement of the magnetic bead subjected to a force (F) induced by a magnetic field gradient [44]. Helicase-catalyzed DNA unwinding results in a stretched single-strand (molecule extension (L)) which enables the magnetic bead to migrate toward the external magnet away from the immobilized DNA end tethered to the coverslip. Video-tracking is employed to detect and measure migration of the microsphere. (E) Combined dual-trap optical tweezers and SM fluorescence microscopy to simultaneously detect and measure unwinding activity and conformation of a DNA helicase protein site-specifically labeled with a fluorophore(s) [45]. The figure depicts an experimental system with the two indicated microspheres in dual optical traps tethered by a DNA hairpin. The confocal microscope (green) detects the conformation of the fluorescently labeled helicase protein complex actively unwinding the duplex DNA hairpin. Reference [46] provides a detailed protocol describing this technique dubbed high-resolution “fleezers”.


Figure 3.
Replication fork structures acted upon by DNA helicases. (A) WRN [72] (or BLM [73,74]) unwind 5’ flaps thereby promoting strand displacement DNA synthesis. In addition, WRN [99,100,101] or BLM [74,102] interacts with FEN-1 and stimulates its 5’ flap endonuclease activity to aid in Okazaki fragment processing. (B) WRN or BLM collaborates with FANCJ to resolve G4 structures that impede DNA synthesis [84]. (C) Pif1 resolves G4 structures on the lagging strand template to allow smooth DNA synthesis [97]. Note: this depiction does not preclude a role of Pif1 in certain contexts to resolve G4 in the leading strand template. (D) RECQL1 reverse branch-migrates regressed fork to restore the replication fork in a manner that is negatively affected by PARP1 [103]. (E) DNA2 nucleolytically processes reversed forks in a process aided by WRN helicase to promote fork restart [104]. (F) The replication fork stalls due to DNA damage or replication stress. FANCJ promotes fork elongation by a mechanism not fully characterized. The fork remodeling factor (not shown) may oppose FANCJ’s role in elongation [105]. (G) E. coli RecG [106] or human WRN [107,108]/BLM [107,109] unwind lagging strand duplex to initiate fork regression when fork stalls. Note: For simplicity, not all fork-interacting proteins are shown in panels. Also, these panels are meant to be representative of DNA structures found at the fork. Not all DNA structures or helicases are shown.
Figure 3.
Replication fork structures acted upon by DNA helicases. (A) WRN [72] (or BLM [73,74]) unwind 5’ flaps thereby promoting strand displacement DNA synthesis. In addition, WRN [99,100,101] or BLM [74,102] interacts with FEN-1 and stimulates its 5’ flap endonuclease activity to aid in Okazaki fragment processing. (B) WRN or BLM collaborates with FANCJ to resolve G4 structures that impede DNA synthesis [84]. (C) Pif1 resolves G4 structures on the lagging strand template to allow smooth DNA synthesis [97]. Note: this depiction does not preclude a role of Pif1 in certain contexts to resolve G4 in the leading strand template. (D) RECQL1 reverse branch-migrates regressed fork to restore the replication fork in a manner that is negatively affected by PARP1 [103]. (E) DNA2 nucleolytically processes reversed forks in a process aided by WRN helicase to promote fork restart [104]. (F) The replication fork stalls due to DNA damage or replication stress. FANCJ promotes fork elongation by a mechanism not fully characterized. The fork remodeling factor (not shown) may oppose FANCJ’s role in elongation [105]. (G) E. coli RecG [106] or human WRN [107,108]/BLM [107,109] unwind lagging strand duplex to initiate fork regression when fork stalls. Note: For simplicity, not all fork-interacting proteins are shown in panels. Also, these panels are meant to be representative of DNA structures found at the fork. Not all DNA structures or helicases are shown.


{kind=link}
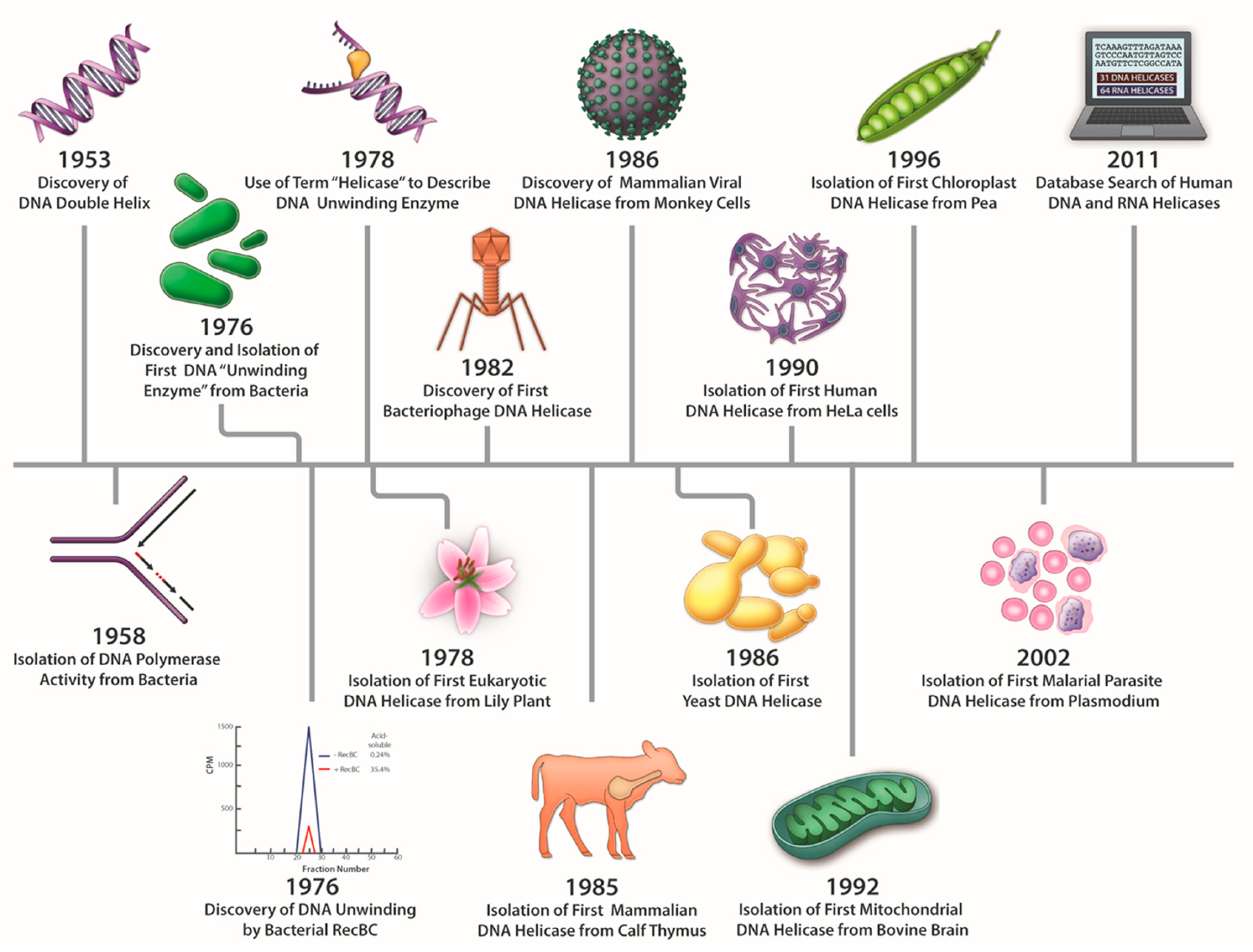{kind=link}
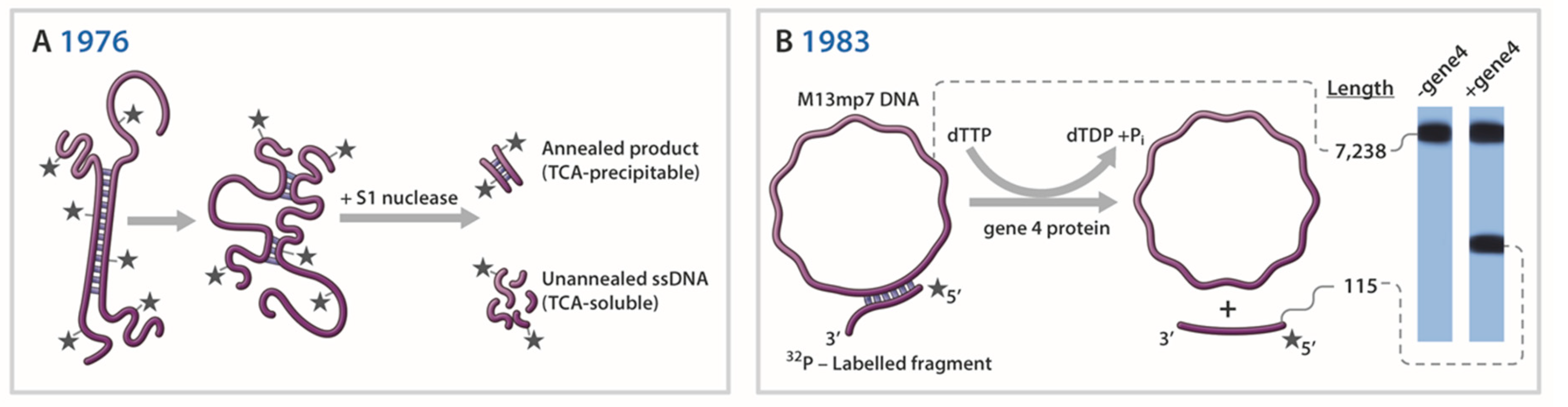{kind=link}
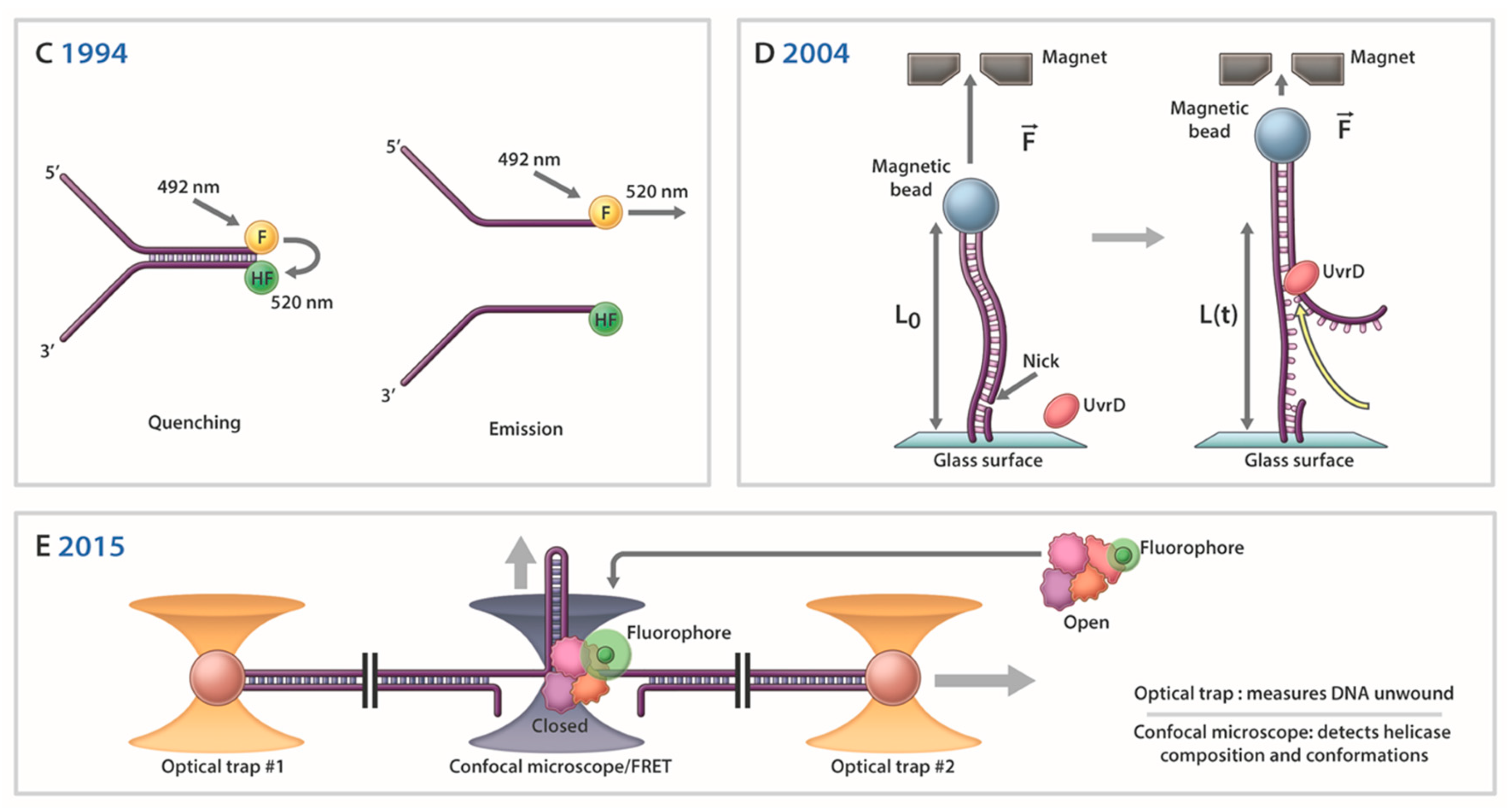{kind=link}
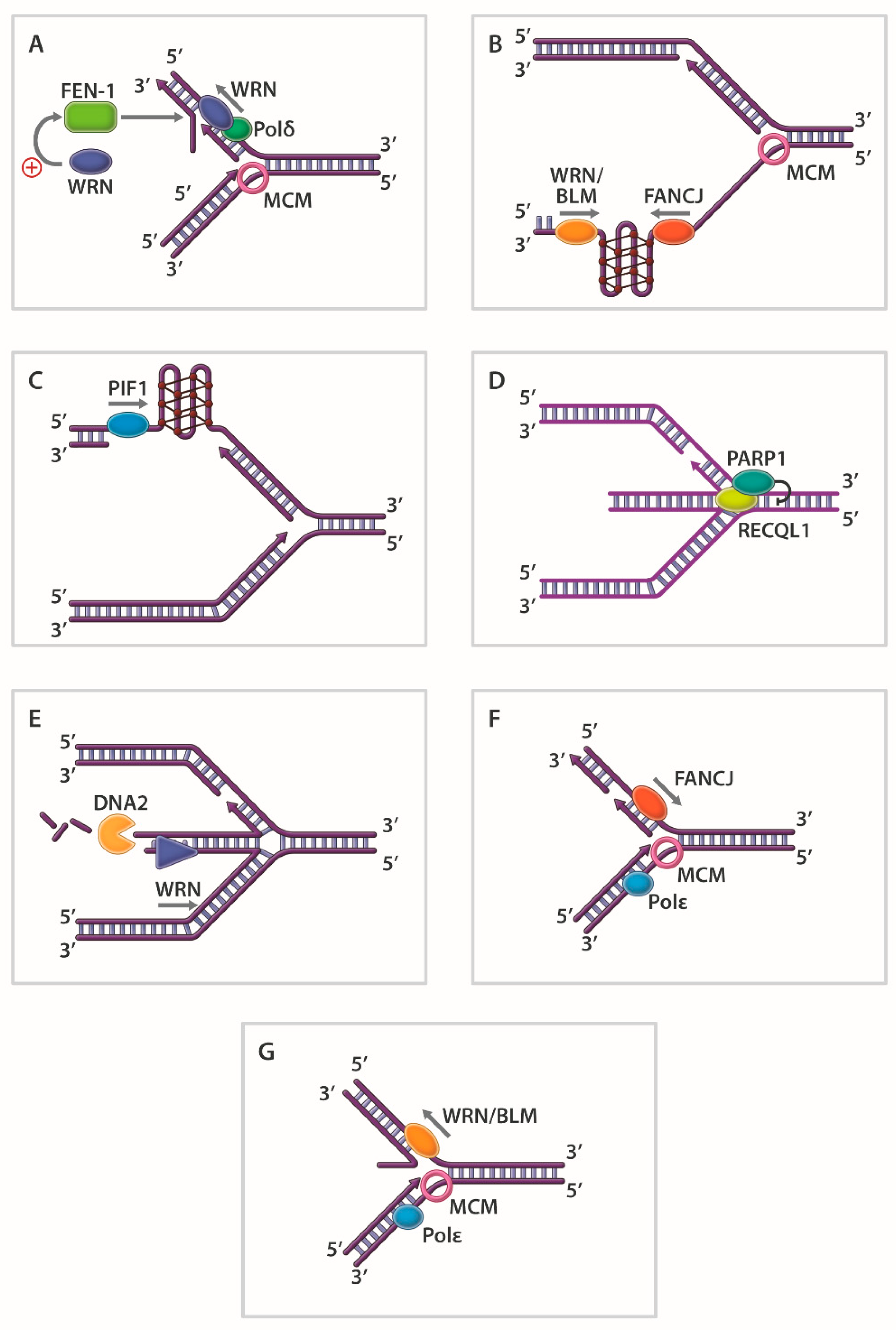{kind=link}
Table 1.
Helicases that Resolve Triplex and G-Quadruplex DNA Structures a.
| Organism/Virus | Resolving Helicase | Reference |
|---|---|---|
| Triplex DNA | ||
| SV40 | large T antigen | [110] |
| H. sapiens | WRN | [111] |
| H. sapiens | BLM | [111] |
| E. coli | RecQ b | [112] |
| H. sapiens | DHX9 | [113,114] |
| H. sapiens | DDX11 (ChlR1) | [115] |
| S. cerevisiae | XPB c | [116] |
| G-quadruplex DNA | ||
| E. coli | RecQ | [117] |
| H. sapiens | BLM | [65,118] |
| S. cerevisiae | Sgs1 | [119] |
| H. sapiens | WRN | [65,120] |
| H. sapiens | FANCJ | [82,83,121] |
| S. cerevisiae | Pif1 | [91] |
| H. sapiens | Pif1 | [92,122] |
| H. sapiens | RTEL1 | [123] |
| H. sapiens | DDX11 (ChlR1) | [124] |
| S. acidocaldarius | XPD | [98] |
a The list is meant to be representative, but not exhaustive, of DNA helicases demonstrated to resolve G4 structures in vitro. b. Triplex resolving activity inferred based on primer extension assays with triplex DNA substrate; see reference for detail. c. TFIIH with ATPase-defective Rad3 was purified from yeast and tested in the assay.
Table 2.
Selected Prominent DNA Helicase Structures a.
| Year | Helicase b | Significant Finding c | Reference |
|---|---|---|---|
| 1989 | SV40 Large T antigen | Double hexamer formed on dsDNA in presence of ATP | [173] |
| 1991 | E. coli rho | Hexamer ring stabilized by RNA d | [183] |
| 1994 | E. coli RuvB | Dodecamer of double hexameric rings around dsDNA | [184] |
| 1995 | T7 gene 4 | Hexameric ring-like structure around ssDNA | [174] |
| 1996 | B. stearothermophilus PcrA | First helicase crystal structure; two RecA-like domains with cleft for nucleotide binding | [50] |
| 1997 | E. coli Rep | First reported crystal structure of helicase bound to DNA | [52] |
| 1999 | T. thermophilus UvrB | Helicase core, DNA and ATP binding domains revealed | [185] |
| 1999 | B. caldotenax UvrB | Structure used to predict UvrB pre-incision complex | [186] |
| 1999 | B. stearothermophilus PcrA | Monomer bound to DNA suggesting an inchworm mechanism | [51] |
| 1999 | T7 gene 4 | Hexamer subunit interactions and nucleotide binding characterized | [187] |
| 2000 | T7 gene 4 | DNA translocation mechanism by sequential nucleotide hydrolysis | [188] |
| 2003 | E. coli RecQ | Conserved Zn2+-binding and winged helix domains in RecQ helicases | [179] |
| 2004 | E. coli RecBCD | Coupling mechanism for motor activities and nuclease shown | [189] |
| 2006 | Papillomavirus E1 | ssDNA passes through ring; escort mechanism for DNA translocation | [190] |
| 2006 | E. coli UvrD | Two-part power stroke (1 bp unwound per ATP hydrolyzed) | [131] |
| 2007 | B. stearothermophilus DnaB | Two-layered ring of hexameric DnaB bound to DnaG Primase | [175] |
| 2008 | S. tokodaii XPD | Two RecA-like domains, Fe-S cluster, and Arch domain revealed | [191] |
| 2008 | S. acidocaldarius XPD | Conserved RecA, Fe-S cluster and Arch domains identified | [192] |
| 2008 | T. acidophilum XPD | Fe-S domain contributes to donut shape for strand separation | [193] |
| 2008 | S. solfataricus MCM | 6-fold symmetry of hexamer; nucleotide-binding pocket at interface | [176] |
| 2009 | D. radiodurans RecD2 | Nucleotide-induced conformational changes; basis for DNA translocation directionality | [53] |
| 2009 | H. sapiens RECQ1 | Winged helix domain harbors β-hairpin strand-separation pin | [194] |
| 2012 | T. acidophilum XPD | Basis for uni-directional ssDNA translocation polarity (5’ -3’) | [54] |
| 2015 | S. cerevisiae Mcm2-7 | Cryo-EM structure suggests narrow passageway fitting duplex DNA | [177] |
| 2015 | H. sapiens BLM | Conformational role of HRDC domain; base-flip unwinding proposed | [195] |
| 2016 | H. sapiens RECQ1 | Strand separation pin buttressed by protein dimer interface | [196] |
| 2016 | S. cerevisiae CMG e | Inchworm mechanism with pumpjack motion for translocation | [197] |
| 2017 | H. sapiens RECQL4 | Novel C-terminal domain not found in other human RecQ helicases | [198] |
| 2017 | H. sapiens RECQL5 | Nucleotide binding dependent open and closed conformations | [199] |
| 2018 | E. coli DinG | Mechanism for unidirectional translocation and lesion stalling | [200] |
| 2018 | S. cerevisiae Pif1 | G4-stabilized dimer formation and a potential G4 binding site | [201] |
| 2018 | C. sakazakii RecQ | RecQ bound to resolved G4; guanine-flipping and sequestration | [202] |
| 2019 | Bacteroides sp. Pif1 | Coordination of two Pif1 molecules for unwinding forked DNA | [203] |
| 2019 | S. solfataricus MCM | DNA translocation mechanism of MCM complex characterized | [172] |
a Chronological list is meant to be representative, but not exhaustive. b Presence of nucleotide and/or DNA is not indicated; see reference for detail. c Major discovery listed; see reference for details of reported findings. d Note: presence of RNA, not DNA. e CMG; Cdc45-Mcm2-7-GINS.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brosh, R.M., Jr.; Matson, S.W. History of DNA Helicases. Genes 2020, 11, 255. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030255
AMA Style
Brosh RM Jr., Matson SW. History of DNA Helicases. Genes. 2020; 11(3):255. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030255
Chicago/Turabian StyleBrosh, Robert M., Jr., and Steven W. Matson. 2020. "History of DNA Helicases" Genes 11, no. 3: 255. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030255
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.