The Increase of Simple Sequence Repeats during Diversification of Marchantiidae, An Early Land Plant Lineage, Leads to the First Known Expansion of Inverted Repeats in the Evolutionarily-Stable Structure of Liverwort Plastomes

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
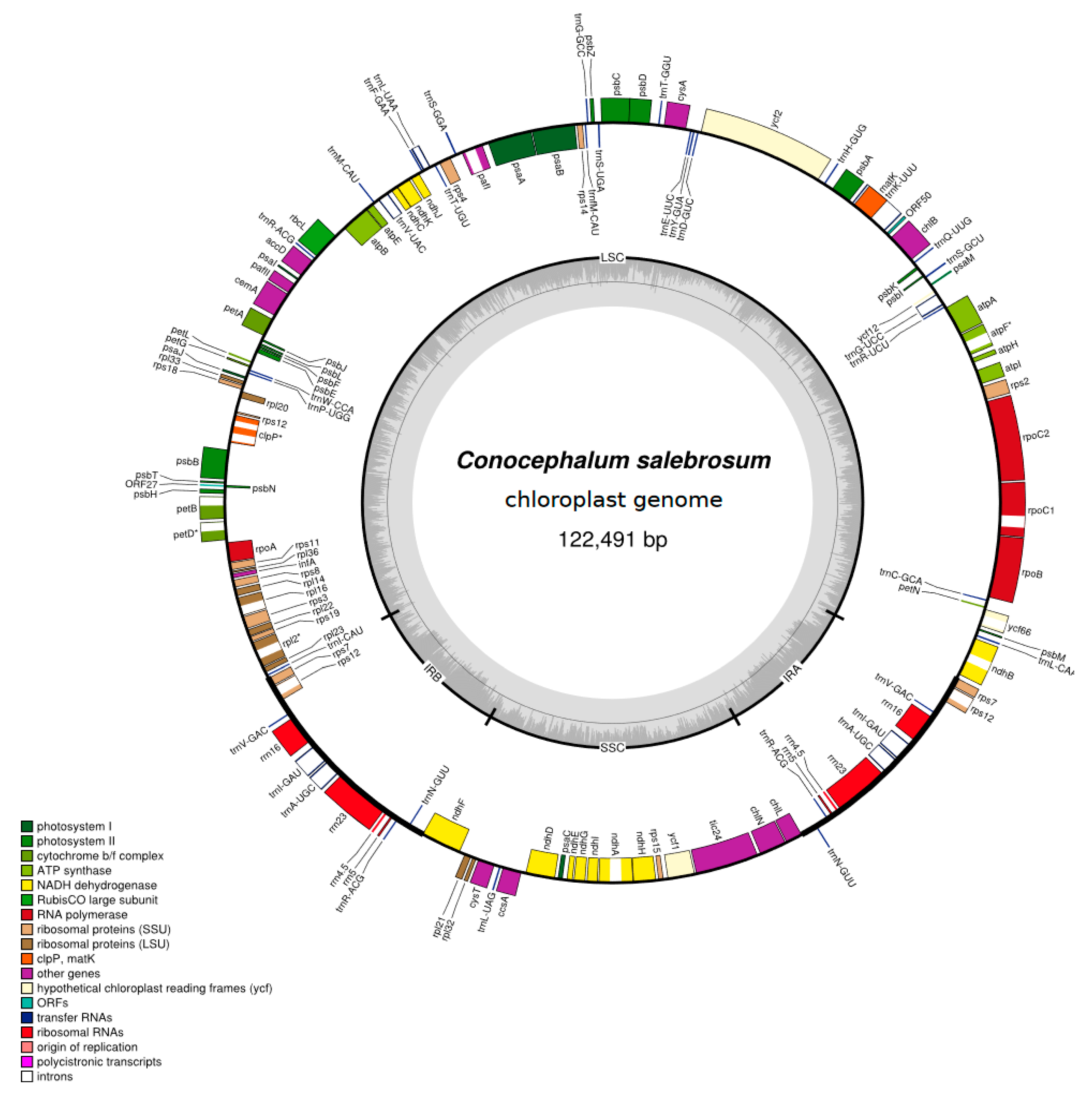
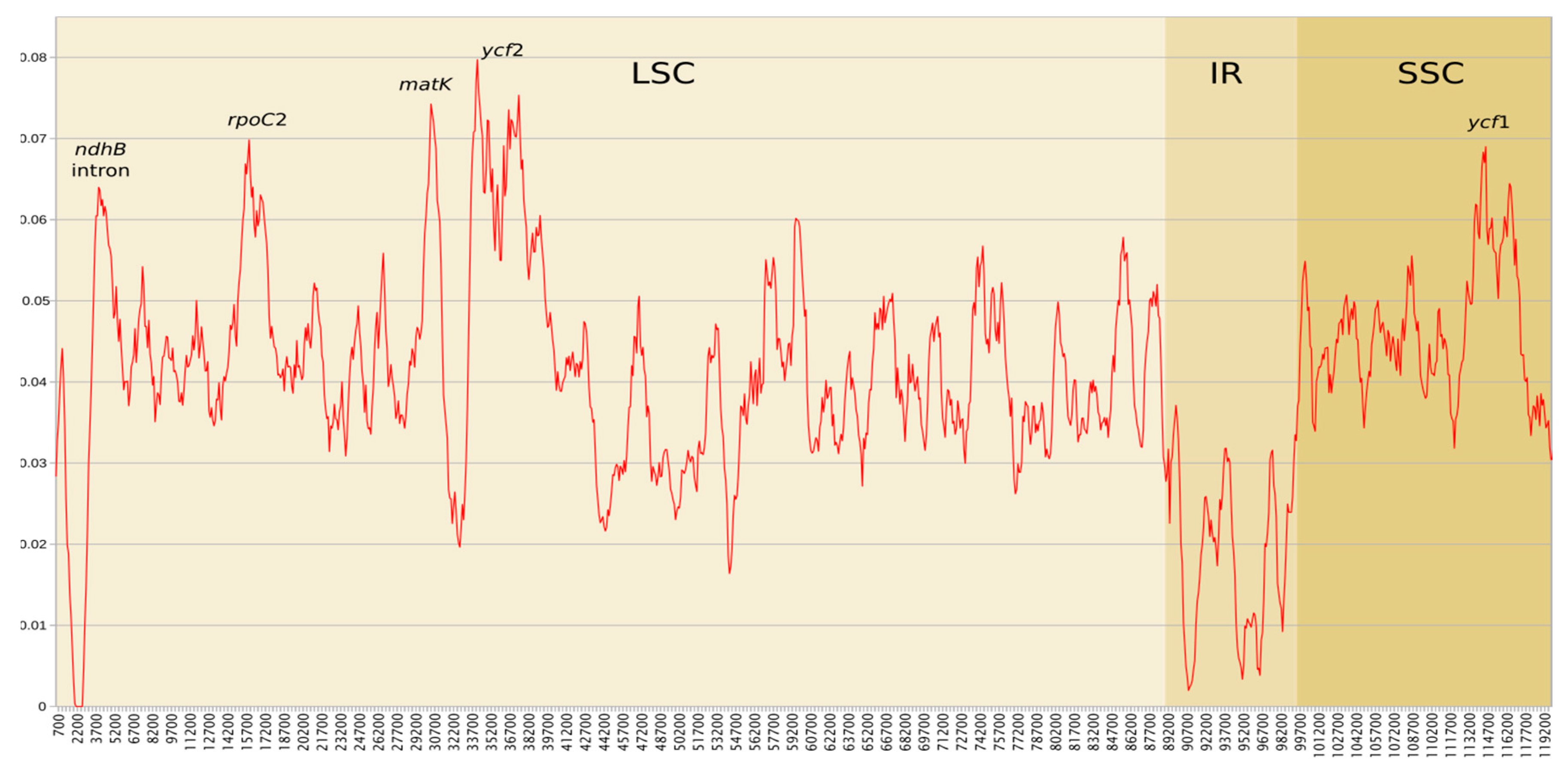
2.1. Characteristics of the Newly Sequenced Chloroplast Genomes
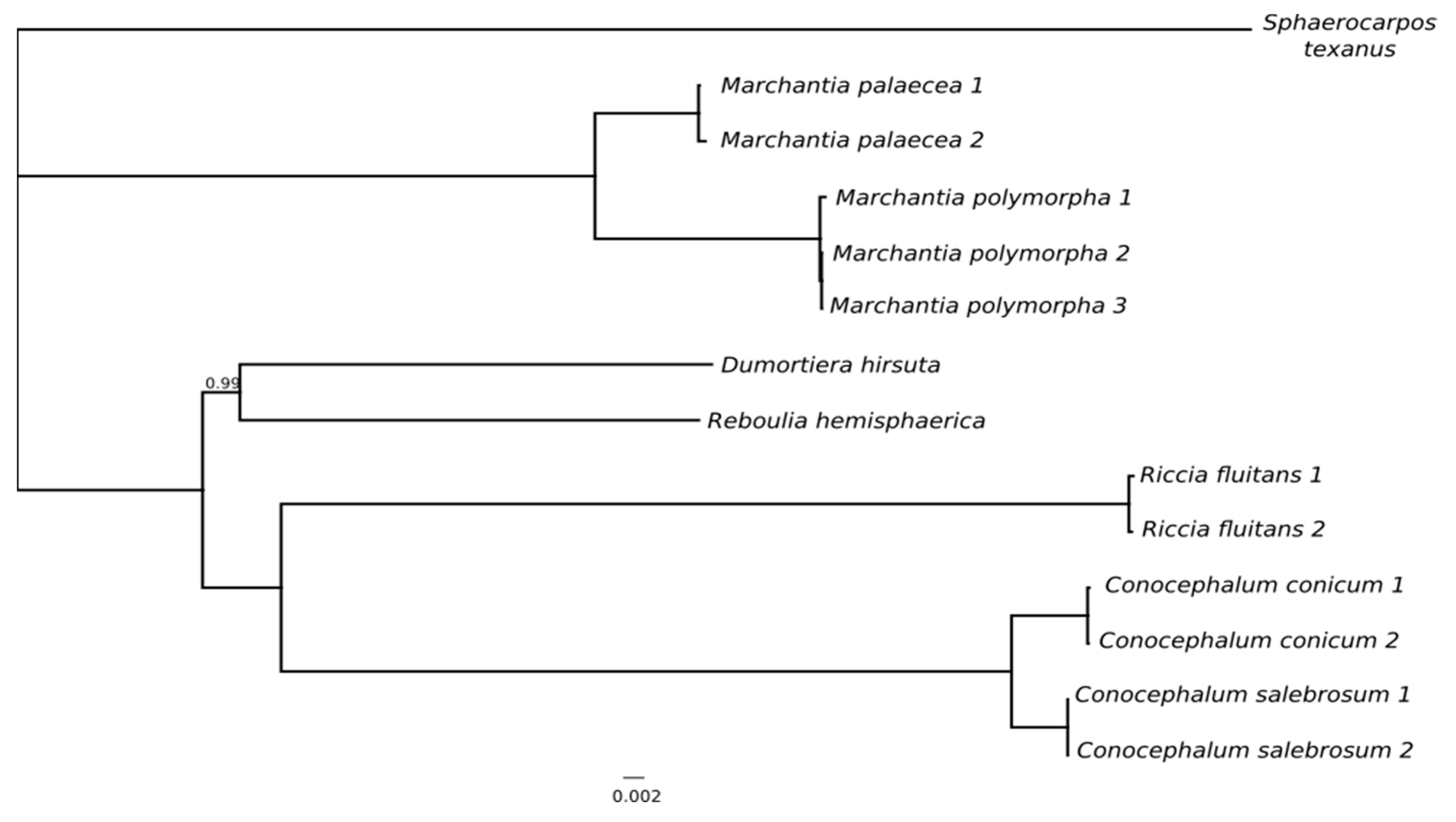
2.2. Phylogenetics Relationships Based on Plastomes Datasets
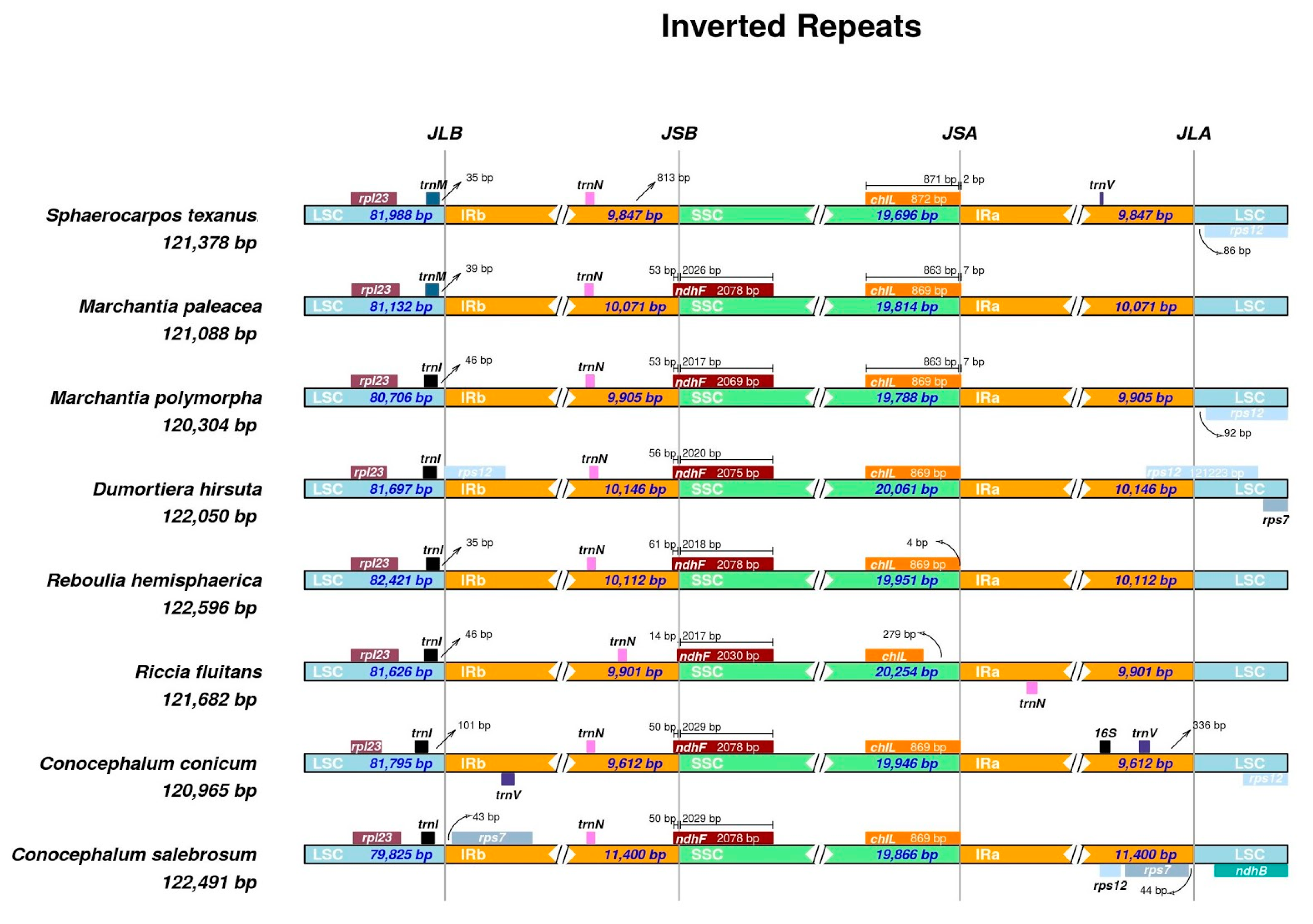
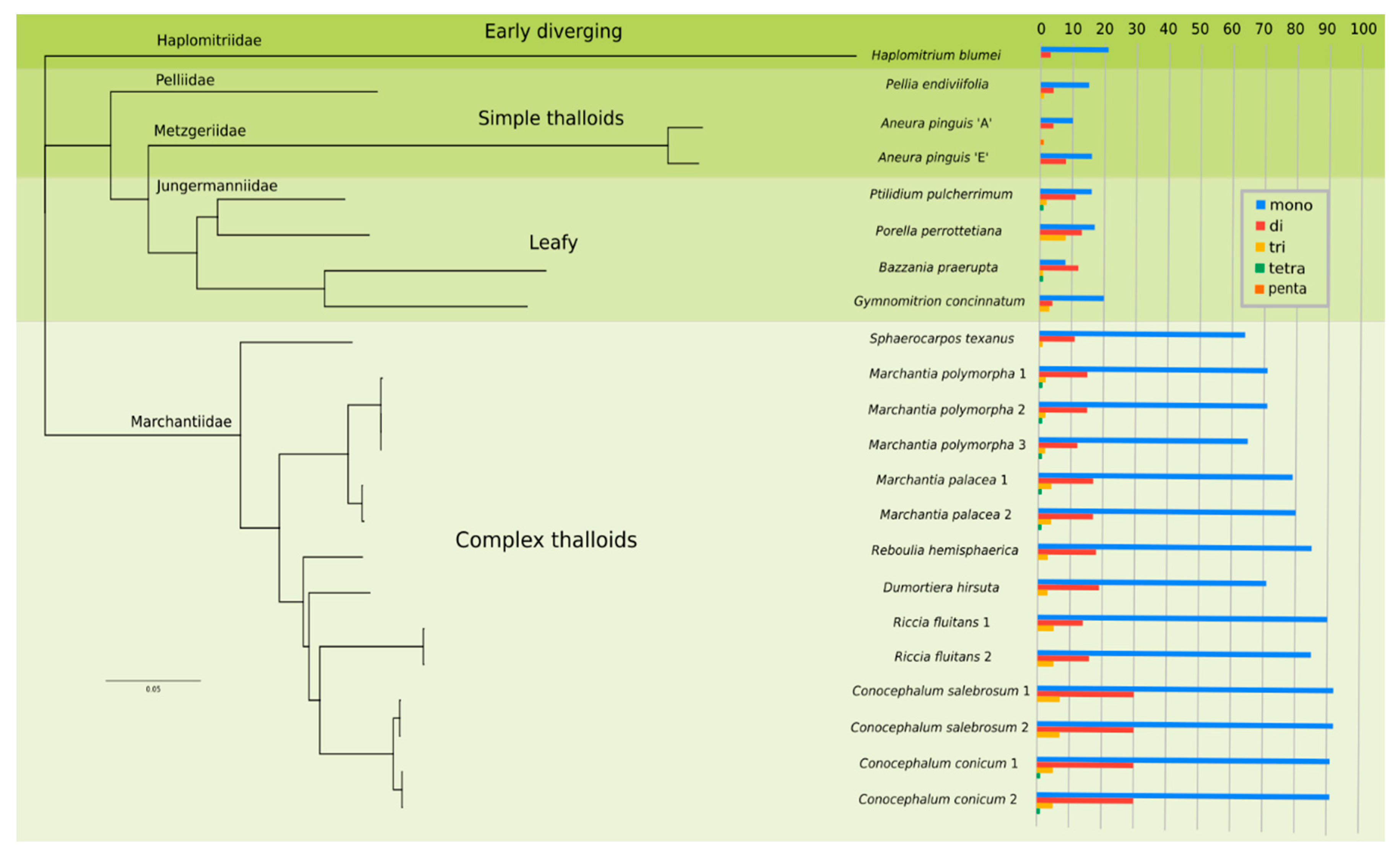
2.3. Mining and Characterisation of SSRs in Liverwort Plastomes
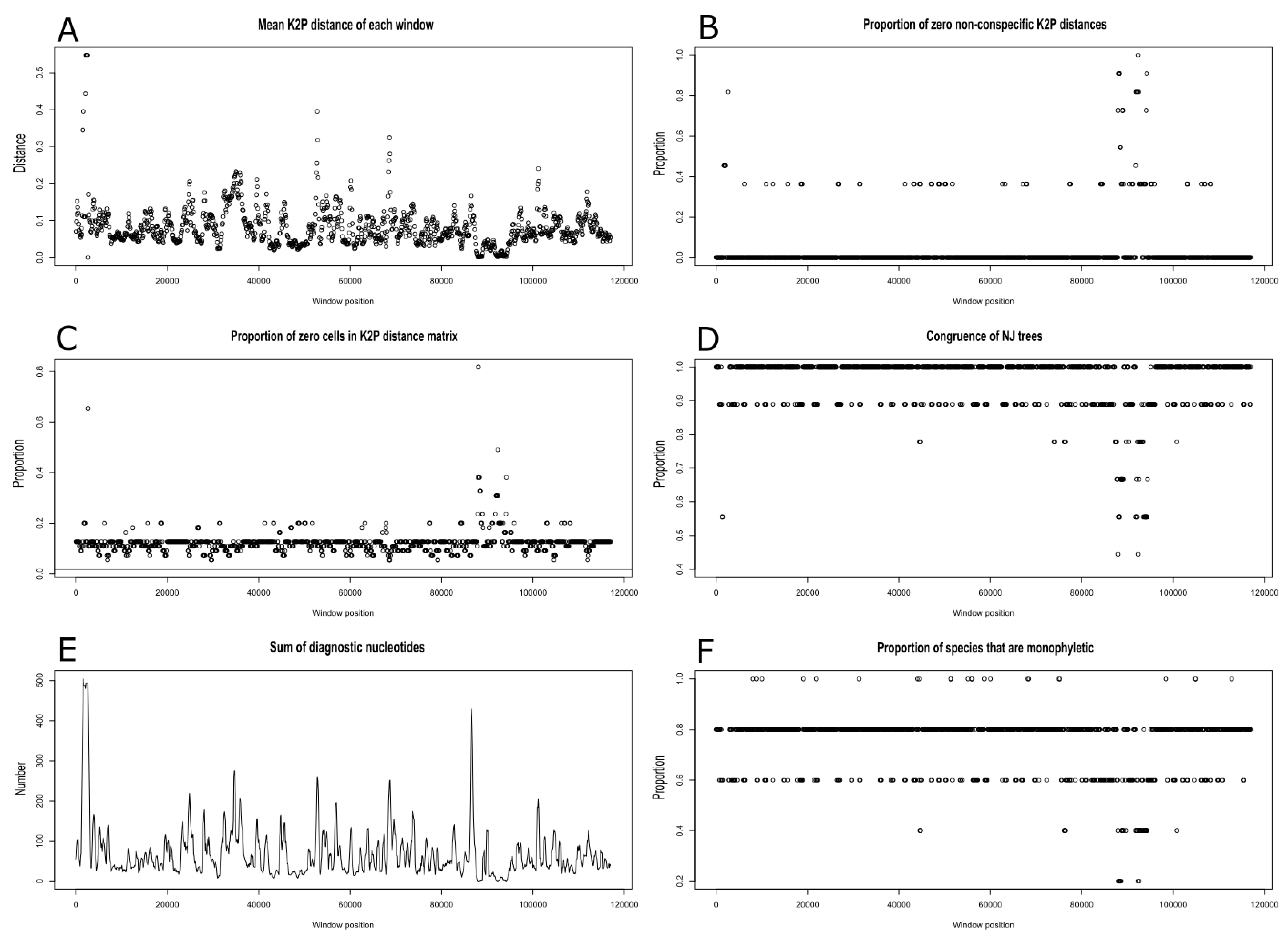
2.4. Plastome Diversity and Molecular Delimitations of Marchantiales
4. Materials and Methods
4.1. DNA Extraction
4.2. Library Construction and Sequencing
4.3. Genomes Assembling, Annotation and Comparative Analyses
4.4. Phylogenomics Analyses
4.5. Diversity Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LSC | Large Singe Copy Region |
| SSC | Small Single Copy Region |
| IR | Inverted Repeats Regions |
| Bp | Base Pair |
| BS | Bootstrap Support |
| SSR | Simple Sequence Repeats |
| ML | Maximum Likelihood |
| MCMC | Monte Carlo Markov Chains |
| NJ | Neighbour-Joining |
References
- Vitelli, M.; Vessella, F.; Cardoni, S.; Pollegioni, P.; Denk, T.; Grimm, G.W.; Simeone, M.C. Phylogeographic structuring of plastome diversity in Mediterranean oaks (Quercus Group Ilex, Fagaceae). Tree Genet. Genomes 2017, 13. [Google Scholar] [CrossRef]
- Huang, B.; Ruess, H.; Liang, Q.; Colleoni, C.; Spooner, D.M. Analyses of 202 plastid genomes elucidate the phylogeny of Solanum section Petota. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Magdy, M.; Ou, L.; Yu, H.; Chen, R.; Zhou, Y.; Hassan, H.; Feng, B.; Taitano, N.; van der Knaap, E.; Zou, X.; et al. Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Sci. Rep. 2019, 8, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Szczecińska, M.; Łazarski, G.; Bilska, K.; Sawicki, J. The complete plastid genome and nuclear genome markers provide molecular evidence for the hybrid origin of pulsatilla × hackelii Pohl. Turk. J. Bot. 2017, 41, 329–337. [Google Scholar] [CrossRef]
- Krawczyk, K.; Nobis, M.; Myszczyński, K.; Klichowska, E.; Sawicki, J. Plastid super-barcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa). Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Szczecińska, M.; Sawicki, J. Genomic resources of three pulsatilla species reveal evolutionary hotspots, species-specific sites and variable plastid structure in the family ranunculaceae. Int. J. Mol. Sci. 2015, 16, 22258–22279. [Google Scholar] [CrossRef] [Green Version]
- Nevill, P.G.; Howell, K.A.; Cross, A.T.; Williams, A.V.; Zhong, X.; Tonti-Filippini, J.; Boykin, L.M.; Dixon, K.W.; Small, I. Plastome-wide rearrangements and gene losses in carnivorous droseraceae. Genome Biol. Evol. 2019, 11, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Forrest, L.L.; Wickett, N.J.; Cox, C.J.; Goffinet, B. Deep sequencing of Ptilidium (Ptilidiaceae) suggests evolutionary stasis in liverwort plastid genome structure. Plant Ecol. Evol. 2011, 144, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Myszczyński, K.; Górski, P.; Ślipiko, M.; Sawicki, J. Sequencing of organellar genomes of Gymnomitrion concinnatum (Jungermanniales) revealed the first exception in the structure and gene order of evolutionary stable liverworts mitogenomes. BMC Plant Biol. 2018, 18, 1–12. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, H.M.; Yang, J.B.; Ma, W.Z.; Pressel, S.; Wu, Y.H.; Schneider, H. Exploring the plastid genome disparity of liverworts. J. Syst. Evol. 2019, 57, 382–394. [Google Scholar] [CrossRef]
- Villarreal A., J.C.; Crandall-Stotler, B.J.; Hart, M.L.; Long, D.G.; Forrest, L.L. Divergence times and the evolution of morphological complexity in an early land plant lineage (Marchantiopsida) with a slow molecular rate. New Phytol. 2016, 209, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Park, H.; Lee, H.; Lee, B.H.; Lee, J. The complete plastome sequence of an antarctic bryophyte Sanionia uncinata (Hedw.) loeske. Int. J. Mol. Sci. 2018, 19, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, C.; Kobayashi, Y.; Aoki, S.; Sugita, C.; Sugita, M. Complete chloroplast DNA sequence of the moss Physcomitrella patens: Evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 2003, 31, 5324–5331. [Google Scholar] [CrossRef]
- Plunkett, G.M.; Downie, S.R. Expansion and Contraction of the Chloroplast Inverted Repeat in Apiaceae Subfamily Apioideae. Syst. Bot. 2000, 25, 648. [Google Scholar] [CrossRef]
- Markham, K.R.; Porter, L.J.; Mues, R.; Zinsmeister, H.D.; Brehm, B.G. Flavonoid variation in the liverwort Conocephalum conicum: Evidence for geographic races. Phytochemistry 1976, 15, 147–150. [Google Scholar] [CrossRef]
- Szweykowski, J.; Krzakowa, M. Variation of four enzyme systems in Polish populations of Conocephalum conicum (L) Dumort (Hepaticae Marchantiales). Bull. Acad. Pol. Sci., Ser. Sci. Biol. 1979, 27, 37–41. [Google Scholar]
- Szweykowski, J.; Buczkowska, K.; Odrzykoski, I.J. Conocephalum salebrosum (Marchantiopsida, Conocephalaceae)—A new Holarctic liverwort species. Plant Syst. Evol. 2005, 253, 133–158. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, C.; Zhang, S.; Wu, H.; Mu, W.; Wei, T.; Li, N.; Wan, T.; Liu, H.; Cui, J.; et al. The Amount of RNA Editing Sites in Liverwort Organellar Genes Is Correlated with GC Content and Nuclear PPR Protein Diversity. Genome Biol. Evol. 2019, 11, 3233–3239. [Google Scholar] [CrossRef]
- Myszczyński, K.; Ślipiko, M.; Sawicki, J. Potential of transcript editing across mitogenomes of early land plants shows novel and familiar trends. Int. J. Mol. Sci. 2019, 20, 2963. [Google Scholar] [CrossRef] [Green Version]
- Myszczyński, K.; Bączkiewicz, A.; Buczkowska, K.; Ślipiko, M.; Szczecińska, M.; Sawicki, J. The extraordinary variation of the organellar genomes of the Aneura pinguis revealed advanced cryptic speciation of the early land plants. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M.; Chang, Z.; et al. Chloroplast gene organization deduced from complete sequence of liverwort marchantia polymorpha chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- George, B.; Bhatt, B.S.; Awasthi, M.; George, B.; Singh, A.K. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 2015, 61, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Brandström, M.; Bagshaw, A.T.; Gemmell, N.J.; Ellegren, H. The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Mol. Biol. Evol. 2008, 25, 2579–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabir, J.S.M.; Yu, M.; Ashworth, M.P.; Baeshen, N.A.; Baeshen, M.N.; Bahieldin, A.; Theriot, E.C.; Jansen, R.K. Conserved gene order and expanded inverted repeats characterize plastid genomes of Thalassiosirales. PLoS ONE 2014, 9, e0107854. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Rabah, S.O.; Shrestha, B.; Hajrah, N.H.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, J.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2019, 57, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yang, J.B.; Ma, W.Z.; Pressel, S.; Liu, H.M.; Wu, Y.H.; Schneider, H. Chloroplast phylogenomics of liverworts: A reappraisal of the backbone phylogeny of liverworts with emphasis on Ptilidiales. Cladistics 2019, 1–10. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, Y.; Choi, C. Chloroplast genome-based hypervariable markers for rapid authentication of six Korean pyropia species. Diversity 2019, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Mizia, P.; Myszczyński, K.; Ślipiko, M.; Krawczyk, K.; Plášek, V.; Szczecińska, M.; Sawicki, J. Comparative plastomes analysis reveals the first infrageneric evolutionary hotspots of orthotrichum s.l. (Orthotrichaceae, Bryophyta). Turk. J. Bot. 2019, 43, 444–457. [Google Scholar] [CrossRef]
- Somaratne, Y.; Guan, D.L.; Abbood, N.N.; Zhao, L.; Wang, W.Q.; Xu, S.Q. Comparison of the complete eragrostis pilosa chloroplast genome with its relatives in eragrostideae (Chloridoideae; poaceae). Plants 2019, 8, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Yang, G.; Peng, J.; Chen, S.; Xu, Z. Determination of the evolutionary pressure on Camellia oleifera on Hainan Island using the complete chloroplast genome sequence. PeerJ 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Logacheva, M.D.; Penin, A.A.; Samigullin, T.H.; Vallejo-Roman, C.M.; Antonov, A.S. Phylogeny of flowering plants by the chloroplast genome sequences: In search of a “lucky gene”. Biochemistry 2007, 72, 1324–1330. [Google Scholar] [CrossRef]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Van Alphen Stahl, J.; Kelly, L.J.; Wikström, N.; Hollingsworth, P.M.; Duff, R.J.; Hoot, S.B.; et al. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc. 2009, 159, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, K.; Szczecińska, M.; Sawicki, J. Evaluation of 11 single-locus and seven multilocus DNA barcodes in Lamium L. (Lamiaceae). Mol. Ecol. Resour. 2014, 14, 272–285. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e0035071. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, W.; Kim, Y.; Park, J. The complete chloroplast genome of Korean Marchantia polymorpha subsp. ruderalis Bischl. & Boisselier: Low genetic diversity between Korea and Japan. Mitochondrial DNA Part B 2019, 4, 959–960. [Google Scholar] [CrossRef] [Green Version]
- Kwon, W.; Kim, Y.; Park, J. The complete chloroplast genome sequence of Dumortiera hirsuta (Sw.) Nees (Marchantiophyta, Dumortieraceae). Mitochondrial DNA Part B 2019, 4, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Grosche, C.; Funk, H.T.; Maier, U.G.; Zauner, S. The chloroplast genome of Pellia endiviifolia: Gene content, RNA-editing pattern, and the origin of chloroplast editing. Genome Biol. Evol. 2012, 4, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Wickett, N.J.; Zhang, Y.; Hansen, S.K.; Roper, J.M.; Kuehl, J.V.; Plock, S.A.; Wolf, P.G.; Depamphilis, C.W.; Boore, J.L.; Goffinet, B. Functional gene losses occur with minimal size reduction in the plastid genome of the parasitic liverwort Aneura mirabilis. Mol. Biol. Evol. 2008, 25, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Fast construction of FM-index for long sequence reads. Bioinformatics 2014, 30, 3274–3275. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; Holt, J.; McMillan, L.; Jones, C.D. FMLRC: Hybrid long read error correction using an FM-index. BMC Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Szczecińska, M.; Gomolińska, A.; Szkudlarz, P.; Sawicki, J. Plastid and nuclear genomic resources of a relict and endangered plant species: Chamaedaphne calyculata (L.) Moench (Ericaceae). Turk. J. Bot. 2014, 38, 1229–1238. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faircloth, B.C. MSATCOMMANDER: Detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour. 2008, 8, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawicki, J.; Bączkiewicz, A.; Buczkowska, K.; Górski, P.; Krawczyk, K.; Mizia, P.; Myszczyński, K.; Ślipiko, M.; Szczecińska, M. The Increase of Simple Sequence Repeats during Diversification of Marchantiidae, An Early Land Plant Lineage, Leads to the First Known Expansion of Inverted Repeats in the Evolutionarily-Stable Structure of Liverwort Plastomes. Genes 2020, 11, 299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030299
Sawicki J, Bączkiewicz A, Buczkowska K, Górski P, Krawczyk K, Mizia P, Myszczyński K, Ślipiko M, Szczecińska M. The Increase of Simple Sequence Repeats during Diversification of Marchantiidae, An Early Land Plant Lineage, Leads to the First Known Expansion of Inverted Repeats in the Evolutionarily-Stable Structure of Liverwort Plastomes. Genes. 2020; 11(3):299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030299
Chicago/Turabian StyleSawicki, Jakub, Alina Bączkiewicz, Katarzyna Buczkowska, Piotr Górski, Katarzyna Krawczyk, Patryk Mizia, Kamil Myszczyński, Monika Ślipiko, and Monika Szczecińska. 2020. "The Increase of Simple Sequence Repeats during Diversification of Marchantiidae, An Early Land Plant Lineage, Leads to the First Known Expansion of Inverted Repeats in the Evolutionarily-Stable Structure of Liverwort Plastomes" Genes 11, no. 3: 299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030299