Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
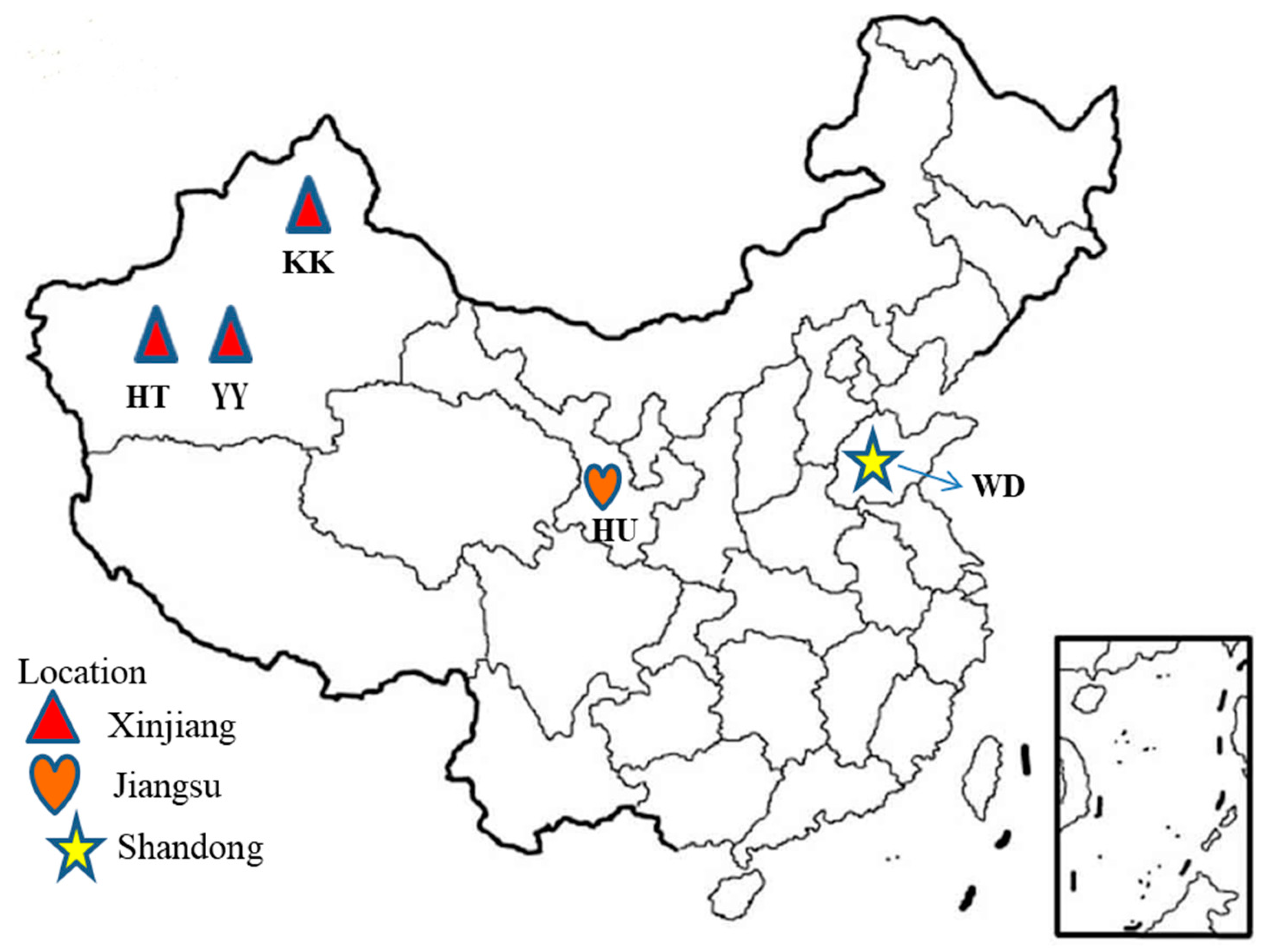
2.2. Samples Collection and DNA Extraction
2.3. Genotyping and Quality Control
2.4. Genomic Diversity and Population Structure Analyses
2.5. Genome-Wide Selection Signatures
2.6. Annotation of Highly Significant Genomic Regions
2.7. Functional Enrichment Analysis of Candidate Genes
3. Results
3.1. Genomic Diversity Analysis
3.2. Population Relationship and Structure Analyses
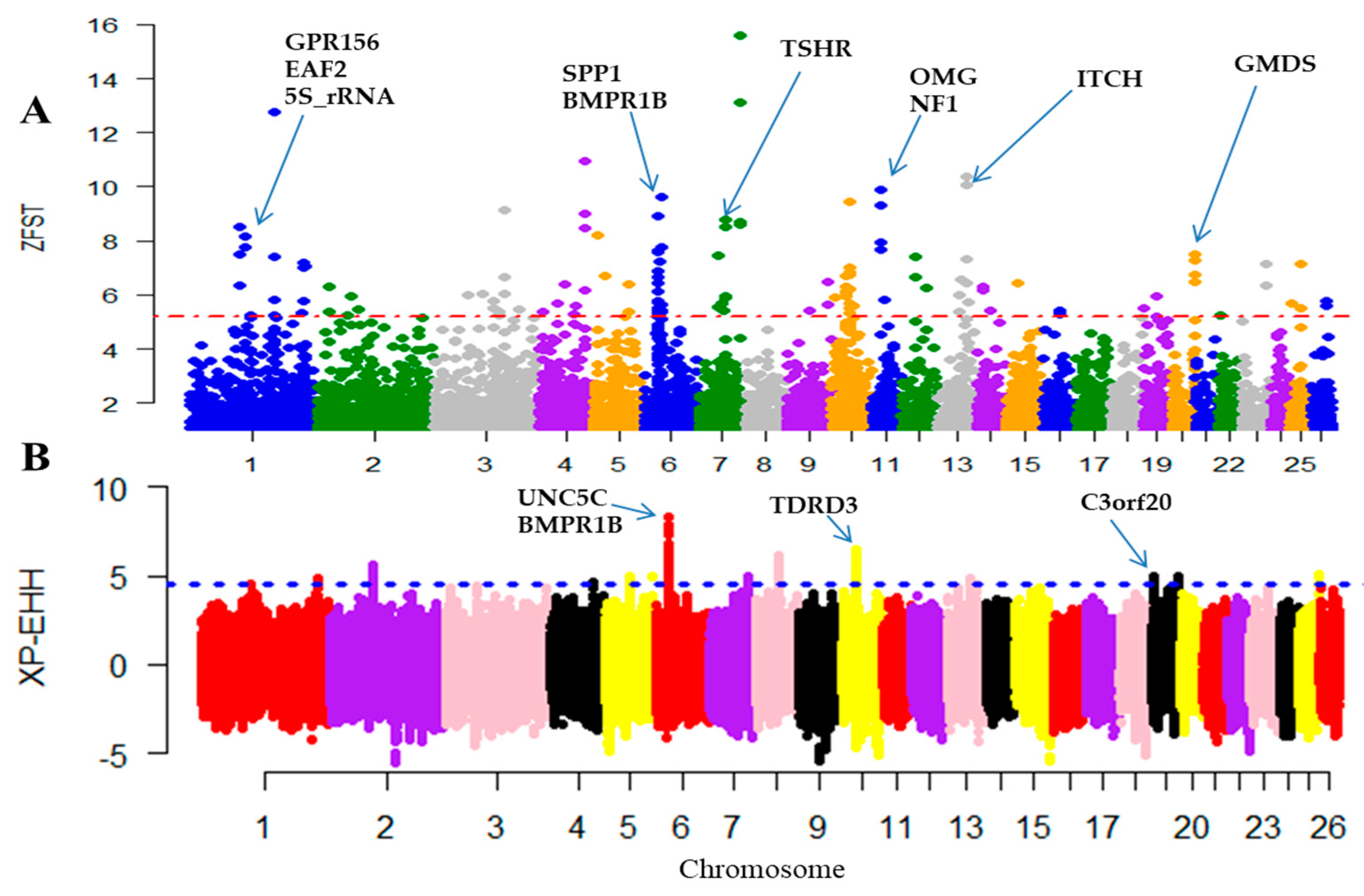
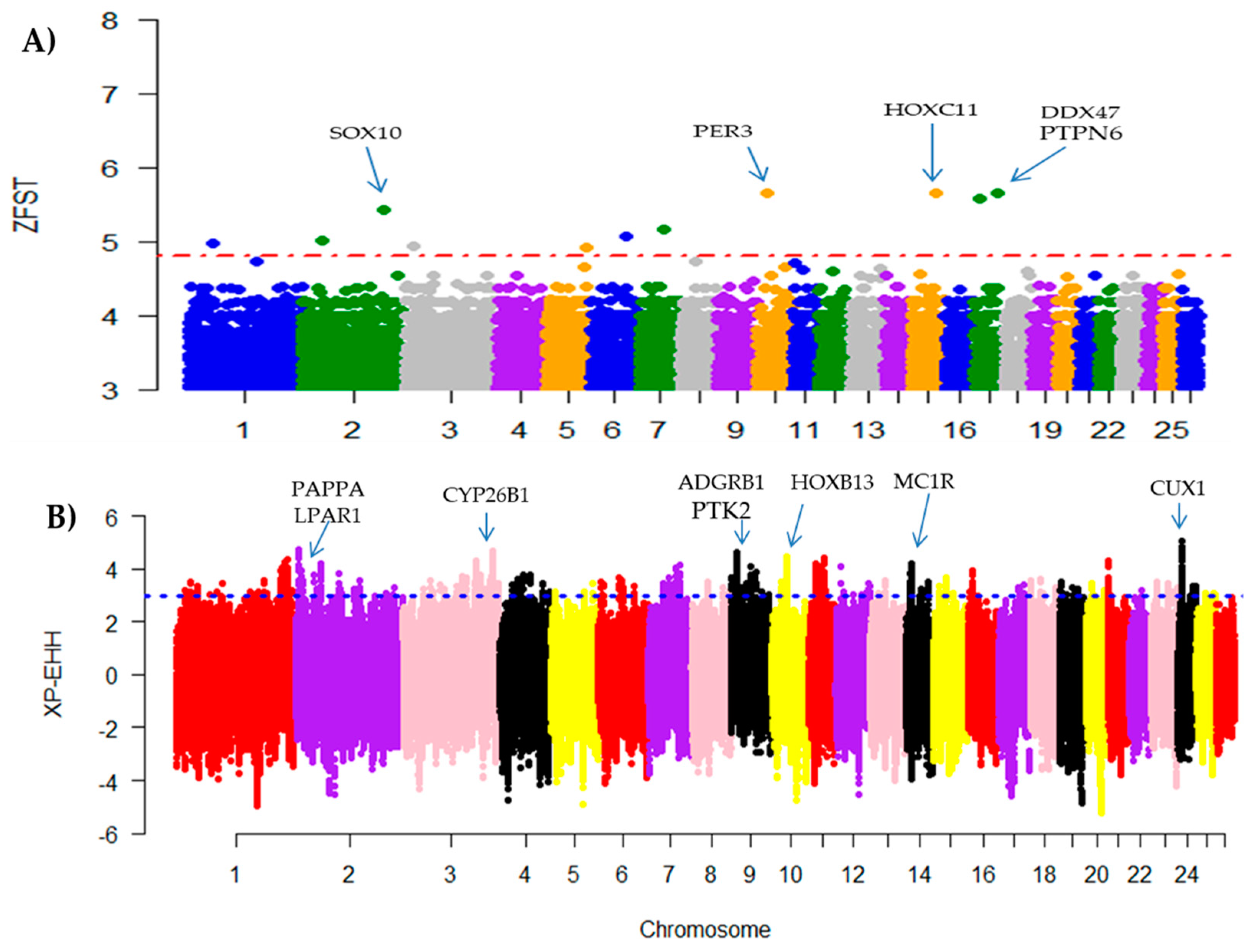
3.3. Detection of Selection Signatures
4. Discussion
4.1. Genomic Diversity within Breeds
4.2. Population Structure of Chinese Local Sheep Breeds from Dry and Humid Environments
4.3. Selection Signatures of Candidate Genes
4.3.1. Candidate Genes Related to Immune Response and Disease Resistance
4.3.2. Candidate Genes Associated with Body Weight and Digestive Metabolism Traits
4.3.3. Candidate Genes Associated to Reproduction Traits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Accessibility
References
- Berman, A. Invited review: Are adaptations present to support dairy cattle productivity in warm climates? J. Dairy Sci. 2011, 94, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, N.R.; Cobanov, B.; Schnitkey, G. Economic losses from heat stress by US livestock industries. J. Dairy Sci. 2003, 86, E52–E77. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, W.-R.; Lv, F.-H.; He, S.-G.; Tian, S.-L.; Peng, W.-F.; Sun, Y.-W.; Zhao, Y.-X.; Tu, X.-L.; Zhang, M. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easterling, D.R.; Meehl, G.A.; Parmesan, C.; Changnon, S.A.; Karl, T.R.; Mearns, L.O. Climate extremes: Observations, modeling, and impacts. Science 2000, 289, 2068–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.-H.; Peng, W.-F.; Yang, J.; Zhao, Y.-X.; Li, W.-R.; Liu, M.-J.; Ma, Y.-H.; Zhao, Q.-J.; Yang, G.-L.; Wang, F. Mitogenomic meta-analysis identifies two phases of migration in the history of eastern Eurasian sheep. Mol. Biol. Evol. 2015, 32, 2515–2533. [Google Scholar] [CrossRef] [Green Version]
- Howden, S.M.; Soussana, J.-F.; Tubiello, F.N.; Chhetri, N.; Dunlop, M.; Meinke, H. Adapting agriculture to climate change. Proc. Natl. Acad. Sci. USA 2007, 104, 19691–19696. [Google Scholar] [CrossRef] [Green Version]
- Lobell, D.B.; Burke, M.B.; Tebaldi, C.; Mastrandrea, M.D.; Falcon, W.P.; Naylor, R.L. Prioritizing climate change adaptation needs for food security in 2030. Science 2008, 319, 607–610. [Google Scholar] [CrossRef]
- Brito, L.F.; Kijas, J.W.; Ventura, R.V.; Sargolzaei, M.; Porto-Neto, L.R.; Cánovas, A.; Feng, Z.; Jafarikia, M.; Schenkel, F.S.J.B.G. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genome 2017, 18, 229. [Google Scholar] [CrossRef] [Green Version]
- Chessa, B.; Pereira, F.; Arnaud, F.; Amorim, A.; Goyache, F.; Mainland, I.; Kao, R.R.; Pemberton, J.M.; Beraldi, D.; Stear, M.J.J.S. Revealing the history of sheep domestication using retrovirus integrations. Science 2009, 324, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Li, S.; Liu, Q.; Wang, Z.; Zhou, Z.; Di, R.; Miao, B.; Hu, W.; Wang, X.; Hu, X. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 2018, 7, giy019. [Google Scholar] [CrossRef]
- Chen, S.Y.; Duan, Z.Y.; Sha, T.; Xiangyu, J.; Wu, S.F.; Zhang, Y.P. Origin, genetic diversity, and population structure of Chinese domestic sheep. Gene 2006, 376, 216–223. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Wu, M.; Cao, J.; Liu, Z.; Liu, R.; Zhao, F.; Zhang, L.; Lu, J.; et al. Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genome 2015, 16, 194. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Li, J.; Ma, N.; Ma, Y.; Wang, J.; Yin, C.; Luo, J.; Liu, N.; Jia, Z.; Fu, C. Animal Genetic Resources in China: Sheep and Goats; China Agriculture Press: Beijing, China, 2011. [Google Scholar]
- Olson, D.M.; Dinerstein, E.; Wikramanayake, E.D.; Burgess, N.D.; Powell, G.V.; Underwood, E.C.; D’amico, J.A.; Itoua, I.; Strand, H.E.; Morrison, J.C.J.B. Terrestrial ecoregions of the world: A New Map of Life on Earth: A new global map of terrestrial ecoregions provides an innovative tool for conserving biodiversity. BioScience 2001, 51, 933–938. [Google Scholar] [CrossRef]
- Karell, P.; Ahola, K.; Karstinen, T.; Valkama, J.; Brommer, J.E. Climate change drives microevolution in a wild bird. Nat. Commun. 2011, 2, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qanbari, S.; Simianer, H.J.L.S. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [Green Version]
- Fariello, M.-I.; Servin, B.; Tosser-Klopp, G.; Rupp, R.; Moreno, C.; San Cristobal, M.; Boitard, S. Selection signatures in worldwide sheep populations. PLoS ONE 2014, 9, e103813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.-S.; Elbeltagy, A.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ji, Z.; Wang, G.; Chao, T.; Hou, L.; Wang, J.J.B. Genome-wide analysis reveals signatures of selection for important traits in domestic sheep from different ecoregions. BMC Genome 2016, 17, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [Green Version]
- De Simoni Gouveia, J.J.; Paiva, S.R.; McManus, C.M.; Caetano, A.R.; Kijas, J.W.; Facó, O.; Azevedo, H.C.; de Araujo, A.M.; de Souza, C.J.H.; Yamagishi, M.E.B.; et al. Genome-wide search for signatures of selection in three major Brazilian locally adapted sheep breeds. Livest. Sci. 2017, 197, 36–45. [Google Scholar] [CrossRef]
- Gutierrez-Gil, B.; Arranz, J.J.; Pong-Wong, R.; García-Gámez, E.; Kijas, J.; Wiener, P. Application of selection mapping to identify genomic regions associated with dairy production in sheep. PLoS ONE 2014, 9, e94623. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B.J.G.S.E. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahbara, A.; Bahbahani, H.; Almathen, F.; Al Abri, M.; Agoub, M.O.; Abeba, A.; Kebede, A.; Musa, H.H.; Mastrangelo, S.; Pilla, F.; et al. Genome-wide variation, candidate regions and genes associated with fat deposition and tail morphology in Ethiopian indigenous sheep. Front. Genet. 2019, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Megdiche, S.; Mastrangelo, S.; Hamouda, M.B.; Lenstra, J.A.; Ciani, E. A combined multi-cohort approach reveals novel and known genome-wide selection signatures for wool traits in Merino and Merino-derived sheep breeds. Front. Genet. 2019, 10, 1025. [Google Scholar] [CrossRef] [Green Version]
- Signer-Hasler, H.; Burren, A.; Ammann, P.; Drögemüller, C.; Flury, C. Runs of homozygosity and signatures of selection: A comparison among eight local Swiss sheep breeds. Anim. Genet. 2019, 50, 512–525. [Google Scholar] [CrossRef]
- Hu, X.-J.; Yang, J.; Xie, X.-L.; Lv, F.-H.; Cao, Y.-H.; Li, W.-R.; Liu, M.-J.; Wang, Y.-T.; Li, J.-Q.; Liu, Y.-G. The genome landscape of Tibetan sheep reveals adaptive introgression from argali and the history of early human settlements on the Qinghai–Tibetan Plateau. Mol. Biol. 2019, 36, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G.J.B. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K.J.G. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.-L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, Å.; Lindblad-Toh, K.J.N. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 2013, 495, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C.J. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Zhi, D.; Da, L.; Liu, M.; Cheng, C.; Zhang, Y.; Wang, X.; Li, X.; Tian, Z.; Yang, Y.; He, T.J. Whole genome sequencing of Hulunbuir short-tailed sheep for identifying candidate genes related to the short-tail phenotype. G3 Genes Genom. Genet. 2018, 8, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.J.N. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Gautier, M.; Vitalis, R.J.B. Rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef]
- Ma, Y.; Ding, X.; Qanbari, S.; Weigend, S.; Zhang, Q.; Simianer, H.J.H. Properties of different selection signature statistics and a new strategy for combining them. Heredity 2015, 115, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Gautier, M.; Klassmann, A.; Vitalis, R. Rehh 2.0: A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Gautier, M.; Naves, M.J.M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011, 20, 3128–3143. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.; Lempicki, R. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A.J.N. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Getachew, T.; Haile, A.; Mészáros, G.; Rischkowsky, B.; Huson, H.J.; Gizaw, S.; Wurzinger, M.; Mwai, A.O.; Sölkner, J. Genetic diversity, population structure and runs of homozygosity in Ethiopian short fat-tailed and Awassi sheep breeds using genome-wide 50k SNP markers. Livest. Sci. 2020, 232, 103899. [Google Scholar] [CrossRef]
- Oliveira, J.C.V.; Ribeiro, M.N.; Rocha, L.L.; Gomes-Filho, M.A.; Delgado, J.V.; Martinez, A.M.; Menezes, M.P.C.; Bettencourt, C.M.; Gama, L.T. Genetic relationships between two homologous goat breeds from Portugal and Brazil assessed by microsatellite markers. Small Rumin. Res. 2010, 93, 79–87. [Google Scholar] [CrossRef]
- Ojango, J.M.; Mpofu, N.; Marshall, K.; Andersson-Eklund, L. Quantitative methods to improve the understanding and utilization of animal genetic resources. Anim. Genet. Train. Resour. 2011, 3, 2011. [Google Scholar]
- Sandenbergh, L.; Cloete, S.W.P.; Roodt-Wilding, R.; Snyman, M.A.; Van der Merwe, A.E. Genetic Diversity and Population Structure of Four South African Sheep Breeds. Proc. Assoc. Advmt. Anim. Breed. Genet 2015, 21, 294–297. [Google Scholar]
- David, C.M.G.; Quirino, C.R.; Vega, W.H.O.; Bartholazzi Junior, A.; Madella-Oliveira, A.F.; Costa, R.L.D. Diversity of indigenous sheep of an isolated population. BMC Vet. Res. 2018, 14, 350. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef]
- Zeng, X.C.; Chen, H.Y.; Hui, W.Q.; Jia, B.; Du, Y.C.; Tian, Y.Z. Genetic diversity measures of 8 local sheep breeds in Northwest of China for genetic resource conservation. Asian Aust. J. Anim. Sci. 2010, 23, 1552–1556. [Google Scholar] [CrossRef]
- Gibbs, R.A. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar] [PubMed] [Green Version]
- Hohenlohe, P.A.; Phillips, P.C.; Cresko, W.A.J.I. Using population genomics to detect selection in natural populations: Key concepts and methodological considerations. Int. J. Plant Sci. 2010, 171, 1059–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Al Kalaldeh, M.; Gibson, J.; Lee, S.H.; Gondro, C.; Van Der Werf, J.H.J. Detection of genomic regions underlying resistance to gastrointestinal parasites in Australian sheep. Genet. Sel. Evol. 2019, 51, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorris, E.R.; Linehan, E.; Trenkmann, M.; Veale, D.J.; Fearon, U.; Wilson, A.G. Association of the rheumatoid arthritis severity variant rs26232 with the invasive activity of synovial fibroblasts. Cells 2019, 8, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorris, E.R.; Tazzyman, S.J.; Moylett, J.; Ramamoorthi, N.; Hackney, J.; Townsend, M.; Muthana, M.; Lewis, M.J.; Pitzalis, C.; Wilson, A.G. The autoimmune susceptibility gene C5orf30 regulates macrophage-mediated resolution of inflammation. J. Immunol. 2019, 202, ji1801155. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Kleine-Eggebrecht, N.; Staufner, C.; Kathemann, S.; Elgizouli, M.; Kopajtich, R.; Prokisch, H.; Lainka, E. Mutation in ITCH gene can cause syndromic multisystem autoimmune disease with acute liver failure. Pediatrics 2019, 143, e20181554. [Google Scholar] [CrossRef] [Green Version]
- Mwacharo, J.M.; Kim, E.-S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.A.; Rothschild, M.F. Genomic footprints of dryland stress adaptation in Egyptian fat-tail sheep and their divergence from East African and western Asia cohorts. Sci. Rep. 2018, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gabel, G.; Aschenbach, J. Influence of food deprivation on the transport of 3-O-methyl-α-D-glucose across the isolated ruminal epithelium of sheep. J. Anim. Sci. 2002, 80, 2740–2746. [Google Scholar] [CrossRef]
- Cormier, A.; Clément, M.-J.; Knossow, M.; Lachkar, S.; Savarin, P.; Toma, F.; Sobel, A.; Gigant, B.; Curmi, P.A. The PN2-3 domain of centrosomal P4.1-associated protein implements a novel mechanism for tubulin sequestration. J. Biol. Chem. 2009, 284, 6909–6917. [Google Scholar] [CrossRef] [Green Version]
- Laske, C.H.; Teixeira, B.B.M.; Dionello, N.J.L.; Cardoso, F.F. Breeding objectives and economic values for traits of low input family-based beef cattle production system in the State of Rio Grande do Sul. Rev. Bras. De Zootec. 2012, 41, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Nosrati, M.; Asadollahpour Nanaei, H.; Amiri Ghanatsaman, Z.; Esmailizadeh, A. Whole genome sequence analysis to detect signatures of positive selection for high fecundity in sheep. Reprod. Domest. Anim. 2019, 54, 358–364. [Google Scholar] [CrossRef]
- Safari, E.; Fogarty, N.; Gilmour, A.R.; Atkins, K.; Mortimer, S.; Swan, A.; Brien, F.; Greeff, J.; Van der Werf, J. Genetic correlations among and between wool, growth and reproduction traits in Merino sheep. J. Anim. Breed. 2007, 124, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, J.P.; Gregan, S.M.; Mulsant, P.; Mullen, M.; Davis, G.H.; Powell, R.; Galloway, S.M. Mutations in the genes for oocyte-derived growth factors GDF9 and BMP15 are associated with both increased ovulation rate and sterility in Cambridge and Belclare sheep (Ovis aries). Biol. Reprod. 2004, 70, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yu, H.; Zhao, X.; Cai, B.; Ding, Q.; Huang, Y.; Li, Y.; Li, Y.; Niu, Y.; Lei, A.J.R. Fertility; Development, Generation of gene-edited sheep with a defined Booroola fecundity gene (FecBB) mutation in bone morphogenetic protein receptor type 1B (BMPR1B) via clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) 9. Repr. Ferti. Dev. 2018, 30, 1616–1621. [Google Scholar]
- Abdoli, R.; Mirhoseini, S.; Ghavi Hossein-Zadeh, N.; Zamani, P.; Gondro, C. Genome-wide association study to identify genomic regions affecting prolificacy in Lori-Bakhtiari sheep. Anim. Genet. 2018, 49, 488–491. [Google Scholar] [CrossRef]
- Abdurahman, A.; Du, X.; Yao, Y.; Sulaiman, Y.; Aniwashi, J.; Li, Q. Smad4 feedback enhances BMPR1B transcription in ovine granulosa cells. Int. J. Mol. Sci. 2019, 20, 2732. [Google Scholar] [CrossRef] [Green Version]
- Shokrollahi, B.; Morammazi, S. Polymorphism of GDF 9 and BMPR 1B genes and their association with litter size in Markhoz goats. Reprod. Domest. Anim. 2018, 53, 971–978. [Google Scholar] [CrossRef]
- Christoforou, E.R.; Pitman, J.L. Intrafollicular growth differentiation factor 9: bone morphogenetic 15 ratio determines litter size in mammals. Biol. Reprod. 2019, 100, 1333–1343. [Google Scholar] [CrossRef]
- Xu, S.-S.; Gao, L.; Xie, X.-L.; Ren, Y.-L.; Shen, Z.-Q.; Wang, F.; Shen, M.; Eyϸórsdóttir, E.; Hallsson, J.H.; Kiseleva, T.J. Genome-wide association analyses highlight the potential for different genetic mechanisms for litter size among sheep breeds. Front. Genet. 2018, 9, 118. [Google Scholar] [CrossRef]
- Bahire, S.V.; Rajput, P.K.; Kumar, V.; Kumar, D.; Kataria, M.; Kumar, S. Quantitative expression of mRNA encoding BMP/SMAD signaling genes in the ovaries of Booroola carrier and non-carrier GMM sheep. Reprod. Domest. Anim. 2019, 54, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, Q.; Di, R.; Hu, W.; Wang, X.; He, X.; Ma, L.; Chu, M. Single nucleotide polymorphisms in BMP2 and BMP7 and the association with litter size in Small Tail Han sheep. Anim. Reprod. Sci. 2019, 204, 183–192. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Code | Location | Sample Size | Ecology | Purpose | PN (%) | HO | HE | FIS | MAF |
|---|---|---|---|---|---|---|---|---|---|---|
| Hetian | HT | Xinjiang | 19 | Arid land | Meat/Wool | 95.5 | 0.367 | 0.360 | −0.02 | 0.269 |
| Karakul | KK | Xinjiang | 20 | Arid/Desert | Lamb skin | 94 | 0.362 | 0.357 | −0.03 | 0.267 |
| Yabuyi | YY | Xinjiang | 20 | Arid | Meat | 93.5 | 0.360 | 0.356 | −0.023 | 0.266 |
| Wadi | WD | Shandong | 17 | Sub-humid | Meat/Wool | 95.3 | 0.360 | 0.360 | −0.004 | 0.270 |
| Hu | HU | Jiangsu | 20 | Humid | Meat/Lamb skin | 95.2 | 0.364 | 0.358 | −0.029 | 0.268 |
| OAR | Gene Position (bp) | Gene Name | Function | Gene Description |
|---|---|---|---|---|
| 1 | 18,323,4435–18,331,6995 | GPR156 | Defenses | G protein-coupled receptor 156 |
| 1 | 18,456,7855–18,461,9155 | EAF2 | Disease resistance | ELL associated factor 2 |
| 1 | 18,334,3776–18,334,3825 | 5S_rRNA | Disease resistance | 5S ribosomal RNA |
| 6 | 36,566,367–36,630,153 | PKD2 | Milk production | Polycystin 2, transient receptor potential cation channel |
| 6 | 36,651,734–36,658,288 | SPP1 | Growth/defense/litter size | Secreted phosphoprotein 1 |
| 6 | 29,361,947–29,448,079 | BMPR1B | Litter size/fecundity/prolifically | Bone morphogenetic protein receptor type 1B |
| 7 | 89,258,424–89,431,877 | TSHR | Reproduction | Thyroid stimulating hormone receptor |
| 11 | 18,317,151–18,318,473 | OMG | Digestive metabolism | Oligodendrocyte myelin glycoprotein |
| 11 | 18,245,395–18,441,418 | NF1 | Litter size | Eurofibromin 1 |
| 13 | 63,388,680–63,446,846 | ITCH | Disease resistance | Itchy E3 ubiquitin protein ligase |
| OAR | Gene Position (bp) | Gene Name | Function | Gene Description |
|---|---|---|---|---|
| 5 | 49,797,770–49,800,184 | PCDHGA2 | Immunity | Protocadherin gamma subfamily A, 2 |
| 5 | 99,509,492–99,557,151 | C5orf30 | Disease resistance | Chromosome 5 open reading frame 30 |
| 6 | 29,146,028–29,348,864 | UNC5C | Reproduction | Unc-5 netrin receptor C |
| 6 | 29,361,947–29,448,079 | BMPR1B | Litter size | Bone morphogenetic protein receptor type 1B |
| 10 | 75,064,027–75,351,616 | DOCK9 | Diseases resistant | Dedicator of cytokinesis 9 |
| 10 | 18,352,91–20,655,11 | TDRD3 | Metabolism | Tudor domain containing 3 |
| 10 | 29,454,677–29,502,617 | RXFP2 | Hair/wool | Relaxin family peptide receptor 2 |
| 13 | 48,462,232–48,472,599 | BMP2 | Fecundity | Bone morphogenetic protein 2 |
| 19 | 57,395,864–57,446,138 | C3orf20 | Adaptive immune | Chromosome 3 open reading frame 20 |
| 25 | 43,127,841–43,132,260 | C10orf71 | Adaptive immune | Chromosome 10 open reading frame 71 |
| OAR | Gene Position (bp) | GENE NAME | Function |
|---|---|---|---|
| 10 | 35,583,842–35,609,414 | FAM117A | Reproduction |
| 10 | 78,274,732–78,283,255 | PER3 | Melatonin and core body temperature rhythms resynchronize |
| 15 | 60,734,22–62,083,71 | HOXC10 | Regulate cell differentiation |
| 15 | 62,437,71–62,716,81 | HOXC11 | Tail fat development |
| 15 | 63,382,71–63,384,20 | HOXC12 | Tail fat development |
| 15 | 63,426,27–64,605,57 | HOXC13 | Tail fat development |
| 15 | 28,607,005–28,679,716 | TBK1 | Innate immune response |
| 15 | 28,718,530–28,746,137 | ZC3H10 | Tail fat development |
| 15 | 28,757,496–287,993,90 | ERBB3 | Normal growth and development |
| 15 | 54,582,673–54,583,137 | CDK2 | Cell cycle regulation, cell proliferation and apoptosis |
| 17 | 22,645,442–22,820,539 | MMP19 | Proliferation |
| 17 | 29,540,625–29,557,094 | SNORA53 | Fat tailed development |
| 17 | 29,631,331–29,661,343 | APAF1 | Immunity |
| 17 | 34,461,385–34,517,413 | DDX47 | Wool Production |
| 17 | 34,971,013–34,971,123 | LRP6 | Disease resistance |
| 17 | 53,661,638–53,752,199 | PHB2 | Immunoprecipitation and embryo developments |
| 17 | 53,803,322–53,859,050 | oar-mir-200c | Milk production |
| 17 | 53,875,546–53,895,506 | PTPN6 | Metabolism |
| 2 | 58,319,311–58,656,152 | LGALS2 | Disease resistance |
| 2 | 10,409,016,7–10,410,456,3 | H1-0 | Antibodies |
| 2 | 10,481,607,5–10,500,749,7 | GCAT | Regulate cardiac development |
| 2 | 17,658,885,2–17,728,788,6 | SOX10 | Coat color patterns |
| 2 | 18,434,313,9–18,434,327,5 | CRPPA | Red meat production |
| 2 | 19,005,983,1–19,006,799,7 | RELN | Fat tailed deposition |
| OAR | Gene Position (bp) | Gene Name | Function |
|---|---|---|---|
| 10 | 35,583,842–35,609,414 | FGF9 | Early gonadal development/testosterone biosynthesis |
| 10 | 35,786,789–35,798,732 | SKA3 | Disease resistance |
| 10 | 35,862,425–35,885,746 | LATS2 | Pubertal development of male reproductive tract and spermatogenesis |
| 10 | 36,045,326–36,103,818 | IFT88 | Reproduction |
| 11 | 15,474,422–15,476,403 | CCL11 | Disease resistance |
| 11 | 36,435,040–36,455,430 | FAM117A | Regulation of ovulation rate |
| 11 | 37,337,231–37,338,988 | HOXB13 | Tail formation |
| 11 | 40,552,263–40,557,934 | KRT27 | Wool follicle and fiber morphology |
| 11 | 46,503,733–46,5282,30 | MRC2 | Immunity |
| 14 | 14,231,363–14,232,541 | MC1R | Black coat color and pigmentation |
| 2 | 72,847,41–75,096,31 | PAPPA | Reproduction (follicle development) |
| 2 | 12,325,968–12,488,187 | LPAR1 | Reproduction (embryo development) |
| 2 | 14,235,503,1–14,244,934,8 | SCN1A | Disease resistance (epilepsy) |
| 2 | 24,675,580,7–24,686,431,8 | PAX7 | Regulation of myogenesis/proliferation |
| 24 | 34,631,867–349,509,62 | CUX1 | Reproduction (Fetal development) |
| 3 | 94,005,022–94,024,647 | CYP26B1 | Regulation of spermatogenesis |
| 3 | 19,093,521,6–19,139,918,6 | SOX5 | Ear developmental processes |
| 3 | 19,337,134,2–193,429,711 | GYS2 | Hepatocyte growth factor |
| 3 | 19,343,685,7–19,344,439,8 | SPX | Antibody response |
| 3 | 20,164,430,6–20,166,437,1 | GSG1 | Fetal development |
| 3 | 20,729,131,1–20,733,999,1 | C1R | Immunity |
| 3 | 20,735,340,8–20,736,655,7 | C1S | Immunity |
| 3 | 20,744,522,8–20,744,919,3 | PHB2 | Embryo development |
| 3 | 20,745,499,6–20,746,386,0 | PTPN6 | Metabolism |
| 3 | 20,758,319,4–20,760,861,0 | CD4 | Immunity (enhances antibody responses) |
| 3 | 20,792,799,7–20,793,488,5 | LTBR | Immune response |
| 3 | 20,798,068,7–20,798,484,4 | TNFRSF1A | Disease resistance |
| 3 | 20,806,906,2–20,808,248,0 | CD9 | Various cellular processes (immune, growth, reproduction). |
| 3 | 20,816,481,5–20,828,841,8 | VWF | Disease resistance |
| 3 | 20,830,234,2–20,864,109,0 | ANO2 | Antibody |
| 9 | 13,460,501–13,464,151 | VPS28 | Growth factor |
| 9 | 14,648,230–14,719,519 | ADGRB1 | Resistance to diarrhea |
| 9 | 15,738,938–15,903,858 | PTK2 | Reproduction |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abied, A.; Bagadi, A.; Bordbar, F.; Pu, Y.; Augustino, S.M.A.; Xue, X.; Xing, F.; Gebreselassie, G.; Han, J.-L.; Mwacharo, J.M.; et al. Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments. Genes 2020, 11, 494. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050494
Abied A, Bagadi A, Bordbar F, Pu Y, Augustino SMA, Xue X, Xing F, Gebreselassie G, Han J-L, Mwacharo JM, et al. Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments. Genes. 2020; 11(5):494. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050494
Chicago/Turabian StyleAbied, Adam, Alnoor Bagadi, Farhad Bordbar, Yabin Pu, Serafino M.A. Augustino, Xianglan Xue, Feng Xing, Gebremedhin Gebreselassie, Jian-Lin Han, Joram M. Mwacharo, and et al. 2020. "Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments" Genes 11, no. 5: 494. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050494