7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
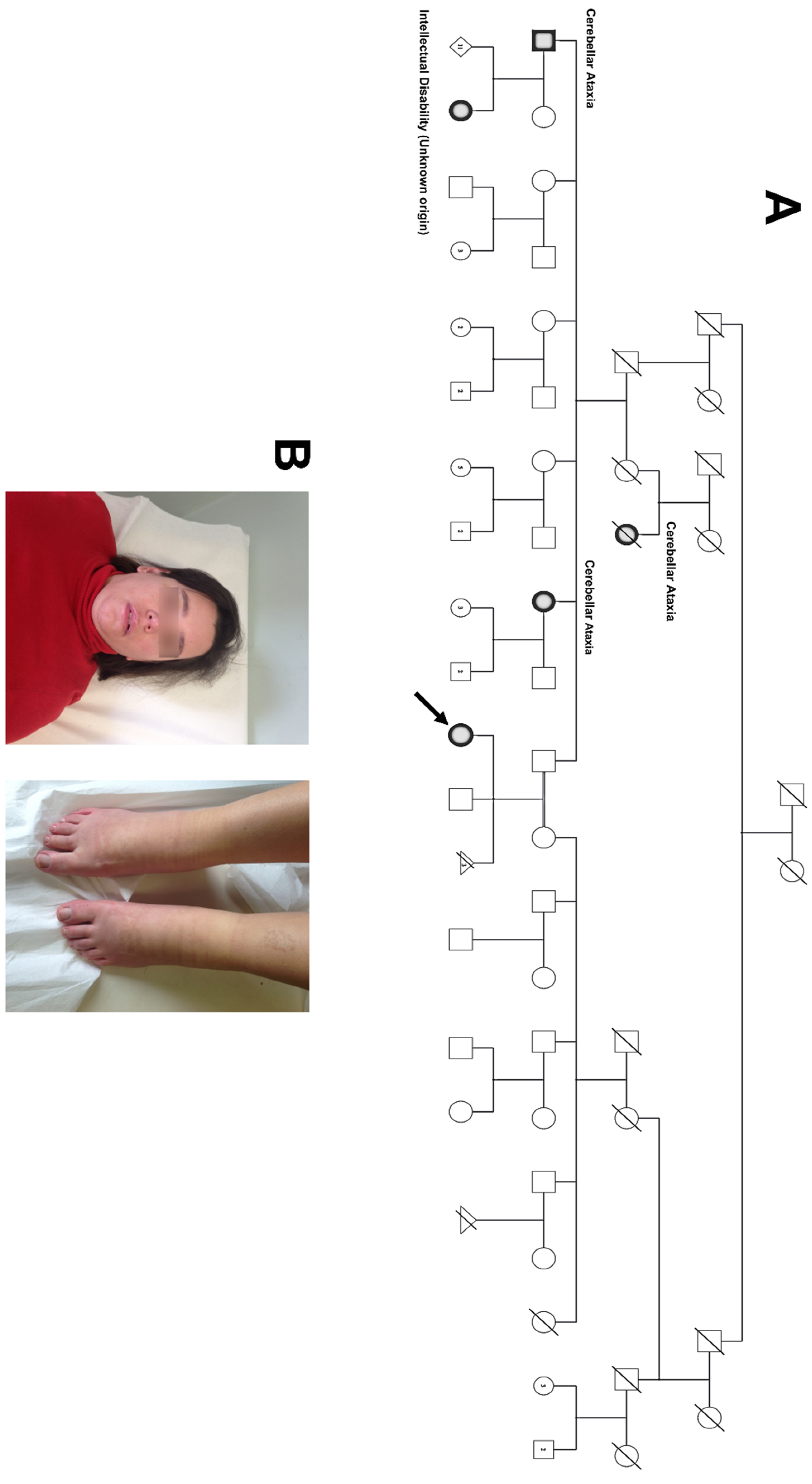
2.1. Proband and Family
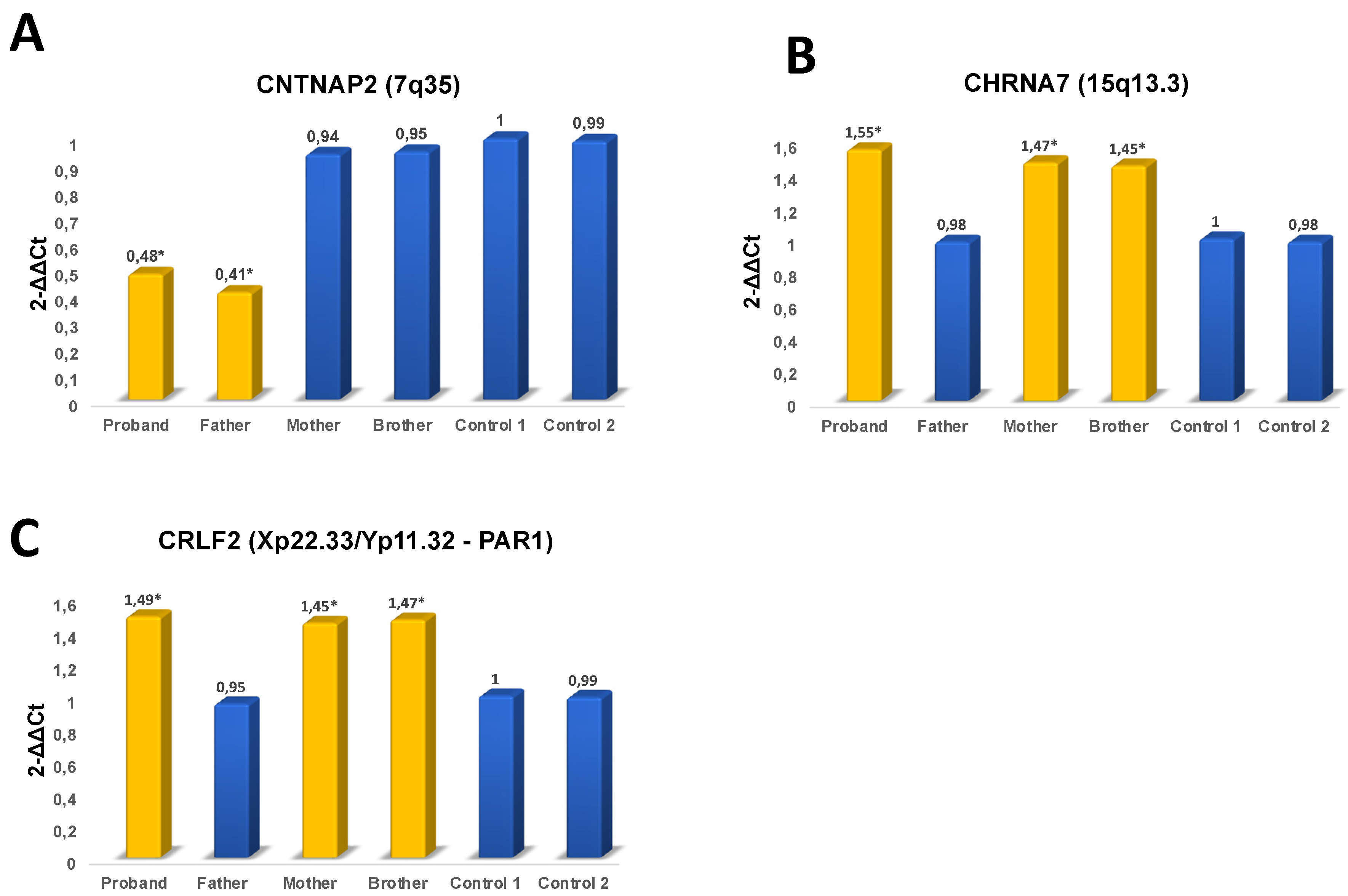
2.2. Molecular Analysis
2.3. Statement of Ethics
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takumi, T.; Tamada, K. CNV biology in neurodevelopmental disorders. Curr. Opin. Neurobiol. 2018, 48, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Merikangas, A.K.; Corvin, A.P.; Gallagher, L. Copy-number variants in neurodevelopmental disorders: Promises and challenges. Trends Genet. 2009, 25, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Hama, Y.; Katsu, M.; Takigawa, I.; Yabe, I.; Matsushima, M.; Takahashi, I.; Katayama, T.; Utsumi, J.; Sasaki, H. Genomic copy number variation analysis in multiple system atrophy. Mol. Brain 2017, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prunier, J.; Caron, S.; MacKay, J. CNVs into the wild: Screening the genomes of conifer trees (Picea spp.) reveals fewer gene copy number variations in hybrids and links to adaptation. BMC Genom. 2017, 18, 97. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-Number variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev.Genom. Human Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [Green Version]
- Consortium, I.S. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237. [Google Scholar]
- Friedman, J.; Vrijenhoek, T.; Markx, S.; Janssen, I.; Van Der Vliet, W.; Faas, B.; Knoers, N.; Cahn, W.; Kahn, R.; Edelmann, L. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 2008, 13, 261. [Google Scholar] [CrossRef]
- Ghani, M.; Pinto, D.; Lee, J.H.; Grinberg, Y.; Sato, C.; Moreno, D.; Scherer, S.W.; Mayeux, R.; George-Hyslop, P.S.; Rogaeva, E. Genome-Wide survey of large rare copy number variants in Alzheimer’s disease among Caribbean hispanics. G3 Genes Genomes Genet. 2012, 2, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Russo, S.; Casula, L.; Alesi, V.; Amendola, F.A.; Angioni, A.; Novelli, A.; Valeri, G.; Menghini, D.; Vicari, S. Array-CGH analysis in a cohort of phenotypically well-Characterized individuals with “essential” autism spectrum disorders. J. Autism Dev. Disord. 2018, 48, 442–449. [Google Scholar] [CrossRef]
- Wincent, J.; Kolbjer, S.; Martin, D.; Luthman, A.; Åmark, P.; Dahlin, M.; Anderlid, B.M. Copy number variations in children with brain malformations and refractory epilepsy. Am. J. Med Genet. Part A 2015, 167, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Gochman, P.; Broadnax, D.D.; Rapoport, J.L.; Ahn, K. 15q13. 3 duplication in two patients with childhood-onset schizophrenia. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2016, 171, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillentine, M.A.; Schaaf, C.P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bon, B.; Mefford, H.; Menten, B.; Koolen, D.; Sharp, A.; Nillesen, W.; Innis, J.; De Ravel, T.; Mercer, C.; Fichera, M. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J. Med Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beal, J.C. Case report: Neuronal migration disorder associated with chromosome 15q13. 3 duplication in a boy with autism and seizures. J. Child Neurol. 2014, 29, NP186–NP188. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.M.; Franke, B.; Mick, E.; Anney, R.J.; Freitag, C.M.; Gill, M.; Thapar, A.; O’Donovan, M.C.; Owen, M.J.; Holmans, P. Genome-Wide analysis of copy number variants in attention deficit hyperactivity disorder: The role of rare variants and duplications at 15q13. 3. Am. J. Psychiatry 2012, 169, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J. Microdeletion/duplication at 15q13. 2q13. 3 among individuals with features of autism and other neuropsychiatric disorders. J. Med Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, K.A.; Reeves, E.; Leavett, R.; Hayiou-Thomas, M.E.; Sharma, A.; Simpson, N.H.; Martinelli, A.; Thompson, P.; Hulme, C.; Snowling, M.J. Copy number variation screen identifies a rare de novo deletion at chromosome 15q13. 1-13.3 in a child with language impairment. PLoS ONE 2015, 10, e0134997. [Google Scholar] [CrossRef]
- Szafranski, P.; Schaaf, C.P.; Person, R.E.; Gibson, I.B.; Xia, Z.; Mahadevan, S.; Wiszniewska, J.; Bacino, C.A.; Lalani, S.; Potocki, L. Structures and molecular mechanisms for common 15q13. 3 microduplications involving CHRNA7: Benign or pathological? Human Mutat. 2010, 31, 840–850. [Google Scholar] [CrossRef] [Green Version]
- Melchior, L.; Bertelsen, B.; Debes, N.M.; Groth, C.; Skov, L.; Mikkelsen, J.D.; Brøndum-Nielsen, K.; Tümer, Z. Microduplication of 15q13. 3 and Xq21. 31 in a family with Tourette syndrome and comorbidities. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2013, 162, 825–831. [Google Scholar] [CrossRef]
- Szatkiewicz, J.P.; O’Dushlaine, C.; Chen, G.; Chambert, K.; Moran, J.L.; Neale, B.M.; Fromer, M.; Ruderfer, D.; Akterin, S.; Bergen, S.E. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 2014, 19, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassfurther, A.; Komini, E.; Fischer, J.; Leipoldt, M. Clinical and genetic heterogeneity of the 15q13. 3 microdeletion syndrome. Mol. Syndromol. 2015, 6, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stobbe, G.; Liu, Y.; Wu, R.; Hudgings, L.H.; Thompson, O.; Hisama, F.M. Diagnostic yield of array comparative genomic hybridization in adults with autism spectrum disorders. Genet. Med. 2014, 16, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tropeano, M.; Howley, D.; Gazzellone, M.J.; Wilson, C.E.; Ahn, J.W.; Stavropoulos, D.J.; Murphy, C.M.; Eis, P.S.; Hatchwell, E.; Dobson, R.J. Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J. Med Genet. 2016, 53, 536–547. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-Time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Human Genet. 2014, 22, 171. [Google Scholar] [CrossRef]
- Poot, M. Intragenic CNTNAP2 deletions: A bridge too far. Mol. Syndromol. 2017, 8, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Toma, C.; Pierce, K.D.; Shaw, A.D.; Heath, A.; Mitchell, P.B.; Schofield, P.R.; Fullerton, J.M. Comprehensive cross-Disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet. 2018, 14, e1007535. [Google Scholar] [CrossRef] [Green Version]
- Poot, M.; Beyer, V.; Schwaab, I.; Damatova, N.; van’t Slot, R.; Prothero, J.; Holder, S.E.; Haaf, T. Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics 2010, 11, 81–89. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Lose, E.J.; Robin, N.H.; Descartes, M.D.; Rutledge, K.D.; Rutledge, S.L.; Korf, B.R.; Carroll, A.J. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med Genet. Part A 2011, 155, 2386–2396. [Google Scholar] [CrossRef]
- Lowther, C.; Costain, G.; Stavropoulos, D.; Melvin, R.; Silversides, C.; Andrade, D. Genetics in Medicine: Official Journal of the American College of Medical Genetics; Nature Publishing Group: New York, NY, USA, 2014. [Google Scholar]
- Cooper, G.; Coe, B.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. Nat. Publ. Group 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll, J.D.; Reuther, G.W. CRLF2 and JAK2 in B-progenitor acute lymphoblastic leukemia: A novel association in oncogenesis. Cancer Res. 2010, 70, 7347–7352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Sharma, J.; Raju, R.; Palapetta, S.M.; Prasad, T.; Huang, T.-C.; Yoda, A.; Tyner, J.W.; Van Bodegom, D.; Weinstock, D.M. TSLP signaling pathway map: A platform for analysis of TSLP-mediated signaling. Database 2014, 2014. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.-H.; de Waal Malefyt, R.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Kitic, M.; Wimmer, I.; Adzemovic, M.; Kögl, N.; Rudel, A.; Lassmann, H.; Bradl, M. Thymic stromal lymphopoietin is expressed in the intact central nervous system and upregulated in the myelin-degenerative central nervous system. Glia 2014, 62, 1066–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, M.R.; Kim, B.S. The itch–scratch cycle: A neuroimmune perspective. Trends Immunol. 2018, 39, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.C.; Mullighan, C.G.; Chen, I.-M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [Green Version]
- Girirajan, S.; Eichler, E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Human Mol. Genet. 2010, 19, R176–R187. [Google Scholar] [CrossRef] [Green Version]
- Gau, S.S.F.; Liao, H.M.; Hong, C.C.; Chien, W.H.; Chen, C.H. Identification of two inherited copy number variants in a male with autism supports two-hit and compound heterozygosity models of autism. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2012, 159, 710–717. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C. A recurrent 16p12. 1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203. [Google Scholar] [CrossRef]
- Rodenas-Cuadrado, P.; Pietrafusa, N.; Francavilla, T.; La Neve, A.; Striano, P.; Vernes, S.C. Characterisation of CASPR2 deficiency disorder-a syndrome involving autism, epilepsy and language impairment. BMC Med Genet. 2016, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paduano, F.; Colao, E.; Loddo, S.; Orlando, V.; Trapasso, F.; Novelli, A.; Perrotti, N.; Iuliano, R. 7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability. Genes 2020, 11, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050525
Paduano F, Colao E, Loddo S, Orlando V, Trapasso F, Novelli A, Perrotti N, Iuliano R. 7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability. Genes. 2020; 11(5):525. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050525
Chicago/Turabian StylePaduano, Francesco, Emma Colao, Sara Loddo, Valeria Orlando, Francesco Trapasso, Antonio Novelli, Nicola Perrotti, and Rodolfo Iuliano. 2020. "7q35 Microdeletion and 15q13.3 and Xp22.33 Microduplications in a Patient with Severe Myoclonic Epilepsy, Microcephaly, Dysmorphisms, Severe Psychomotor Delay and Intellectual Disability" Genes 11, no. 5: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050525