Rapid Evaluation of CRISPR Guides and Donors for Engineering Mice

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blastocysts Assay Overview
2.2. Design and Preparation of Single Guide RNA
2.3. Design and Preparation of Single Strand Donor DNA
2.4. Zygote Preparation, Injection, Culture and Lysis
2.5. Primers
2.6. PCR
2.7. Analysis
ICE
2.8. Mice
3. Results
3.1. General Strategy
3.2. Method Outline
- Design and make or buy guide RNA and donor DNA.
- Design and make or buy guide RNA and donor DNA.
- Design a minimum of 2 sets of optimal primers to perform PCR around the CRISPR cut site and insertion site, one set inside of the other, for eventual use in nested PCR. Center the oligo made by the inner primers on the desired point mutations and include the guide cut region. Each primer for the inner set should be roughly 100 to 200 base pairs away from the closest point mutation or cut point, to give a final product of about 200 to 400 base pairs. This will produce the cleanest, strong, clear Sanger sequence peaks in the region containing the desired mutations to be analyzed. We typically chose outer primers to give a fragment of about 400 to 700 base pairs in size, to save on PCR time.
- Inject Cas9, guide RNA and donor DNA into mouse single cell embryos and incubate for 3 to 3.5 days until they reach the blastocyst stage. We usually injected enough embryos to produce at least 8 blastocysts for each guide/donor pair, though not all blastocyst preparations give sequenceable PCR product. Morulae frequently also give usable results. With a good guide and donor pair producing above 50% donor sequence integration on average, as few as 4 embryos can be used, though we suggest using 8 or more.
- Pick up blastocysts in the smallest volume of culture medium possible and lyse in a maximum of 10 μL final volume of lysing solution under ~10–20 μL mineral oil (to reduce evaporation). We typically used 2.5 μL of Extraction Reagent lysis solution (Quanta Biosciences). The solution of lysed blastocysts was mixed well by flicking and briefly centrifuging (microcentrifuge, repeated once), heated at 95 °C for 30 min, and then neutralized with an equivalent amount of Stabilization Buffer per manufacturer’s instructions. Homemade lysing solution gave similar results.
- Perform PCR with the outer set of primers, using a high-fidelity polymerase as recommended by the manufacturer, except increase elongation time by either 4 times the recommended, or by 2 times and use 60 cycles instead of the usual 30–35 cycles. We ran a 25 μL reaction using Q5 polymerase (New England Biolabs, Ipswich, MA) and 1 μL of the lysed blast mixture. To get consistent results, it is important to mix the lysed blastocyst solution well but gently and spin it down before adding it to the PCR mixture.
- Repeat the PCR with the inner set of primers and 1 μL of the product from the first PCR, again using a high-fidelity polymerase with the same increased elongation times/cycles as the first PCR.
- Run the 2nd PCR product on a 2% agarose gel. If a single band can be seen and is at the appropriate molecular weight, despite possibly being quite faint or including high molecular weight smears, it can be sequenced.
- Sequence PCR products that produce a visible band on the agarose gel. The same primer used in the 2nd PCR and upstream of the desired point mutations rather than upstream of the cut, can also be used for sequencing. To clean the DNA, we typically mixed 3 μL ExoSAP-IT with 7.5 μL of the PCR product, incubated at 37 °C for 5 min and 1 min at 80 °C, then sent the resulting mixture in for sequencing.
- Guide RNA/donor DNA pairs that show visible integration of the desired mutations in more than 50% of the blastocysts are good candidates for making mice with these point mutations. The different pairs can also be compared to choose the best among them. If quantitation of the percent of indel and point mutations in each blastocyst is desired, Synthego’s ICE program works well.
3.3. CRISPR Component Toxicity
3.4. CRISPR Component Leakage
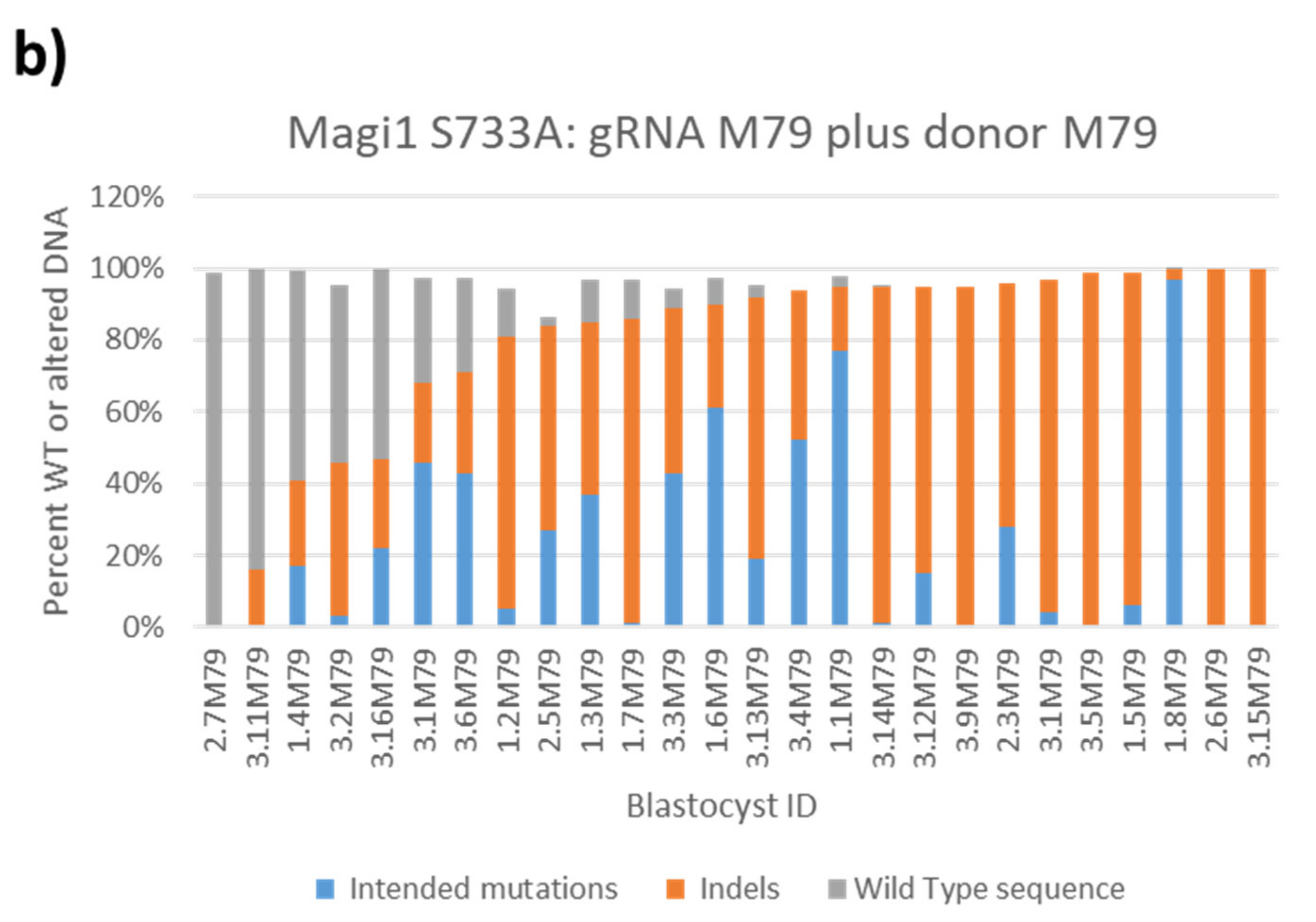
3.5. Repeatable Donor Integration Efficiency with the Same Guide/Donor Pair
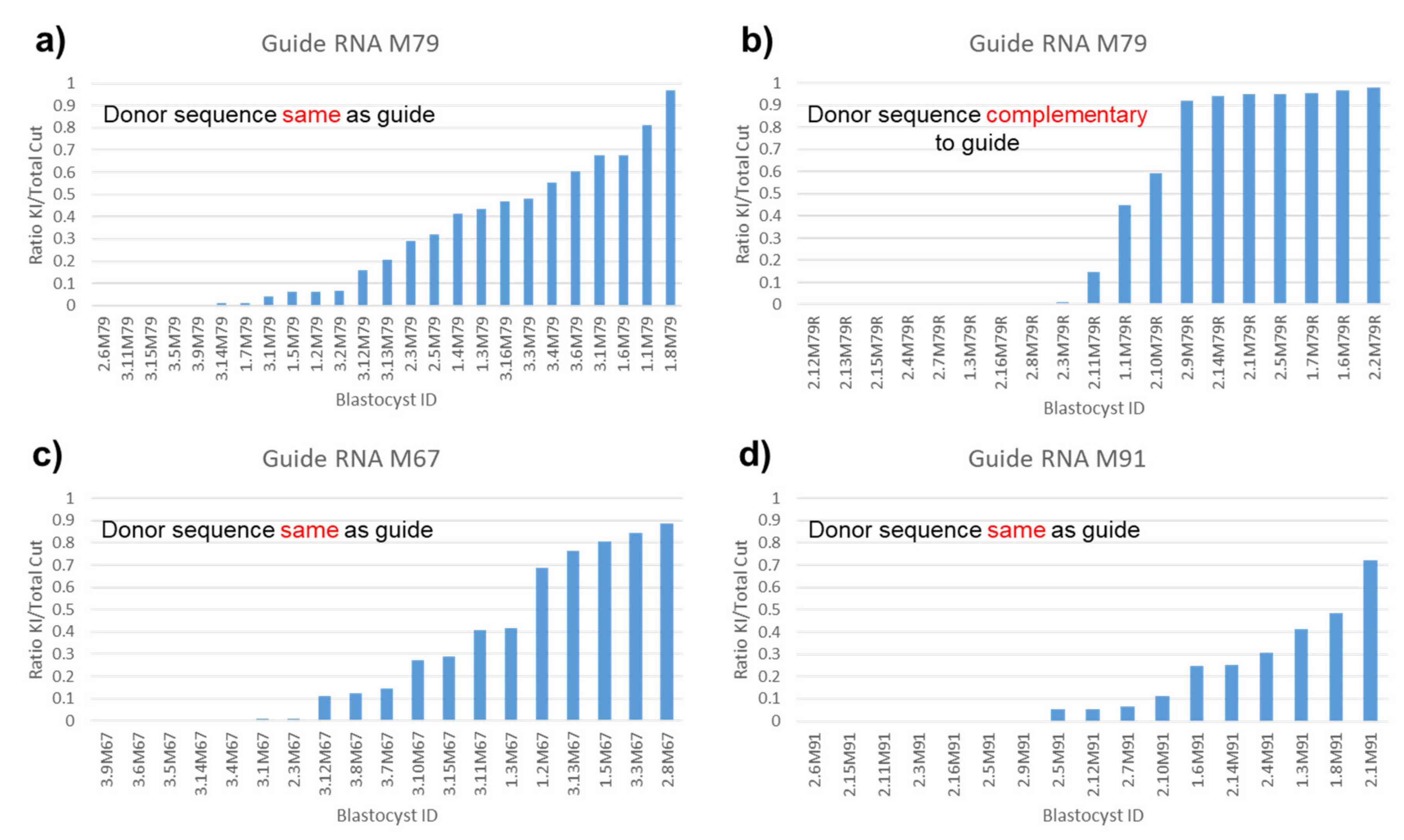
3.6. All-or-None Sequence Integration with Complementary Donor
3.7. Increased Knock-in Efficiency of Distant Mutagenesis Sites with Silent Mutations
3.8. Similar Donor Integration in All Genes Tested
3.9. Unequal Efficiency in Dual Donor Sequence Integration
3.10. Similar Donor Sequence Integration Efficiency in Mice vs. Blastocysts
4. Discussion
4.1. Advantages of the Blastocyst Protocol
4.2. Reproducibility of Blastocyst Protocol
4.3. Similar Pattern of Donor Integration into Different Genes
4.4. Increased Integration Efficiency Using Silent Mutations
4.5. Effect of Donor Direction and Number on Donor Sequence Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Blasts | Blastocysts |
| Cas9 | CRISPR associated protein 9 |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DNP | did not progress to the late morula/blastocyst stage |
| ICE | Inference of CRISPR Edits |
| KI | Knock-in |
| KO | knock-out |
| kb | kilobase |
| Tm | melting temperature |
| oligos | oligonucleotides |
| HDR | homology directed repair |
| ssDNA | single strand DNA |
| sgRNA | single guide RNA |
| WGA | whole genome amplification |
| WT | wildtype |
References
- Behringer, R.; Gertsenstein, M.; Nagy, K.V.; Nagy, A. Manipulating the Mouse Embryo: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2014; ISBN 978-1-936113-01-9. [Google Scholar]
- Xu, H.; Xiao, T.; Chen, C.-H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehravar, M.; Shirazi, A.; Mehrazar, M.M.; Nazari, M. In Vitro Pre-validation of Gene Editing by CRISPR/Cas9 Ribonucleoprotein. Avicenna J. Med. Biotechnol. 2019, 11, 259–263. [Google Scholar] [PubMed]
- Anders, C.; Jinek, M. In vitro Enzymology of Cas9. Methods Enzymol. 2014, 546, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tjian, R.; Weissman, J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife 2016, 5, e12677. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Bray, N.L.; Schaefer, A.J.; Floor, S.; Corn, J. CRISPR-Cas9 Genome Editing In Human Cells Works Via The Fanconi Anemia Pathway. bioRxiv 2017, 136028. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Elliott, B.; Richardson, C.; Winderbaum, J.; Nickoloff, J.A.; Jasin, M. Gene Conversion Tracts from Double-Strand Break Repair in Mammalian Cells. Mol. Cell. Biol. 1998, 18, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Song, F.; Stieger, K. Optimizing the DNA Donor Template for Homology-Directed Repair of Double-Strand Breaks. Mol. Ther. Nucleic Acids 2017, 7, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, W.; Kawasaki, K.; Sugiura, K.; Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 2013, 41, e187. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Watanabe, S.; Kamiyoshi, A.; Sato, M.; Shindo, T. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC Biotechnol. 2014, 14, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.T.; Weber, T.; Graf, R.; Sommermann, T.; Petsch, K.; Sack, U.; Volchkov, P.; Rajewsky, K.; Kühn, R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Zhang, M.; Wang, X.; Ying, W.; Hu, X.; Dai, P.; Meng, F.; Shi, L.; Sun, Y.; Yao, N.; et al. Tild-CRISPR Allows for Efficient and Precise Gene Knockin in Mouse and Human Cells. Dev. Cell 2018, 45, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, J.; Ko, K.A.; Kotla, S.; Wang, Y.; Paez-Mayorga, J.; Shin, I.J.; Imanishi, M.; Vu, H.T.; Tao, Y.; Leiva-Juarez, M.M.; et al. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Shin, I.; Won, J.H.; Ko, K.A.; Shin, J.-H.; McBeath, E.; Thomas, T.; Giancursio, C.; RA, Q.-Q.; Taunton, J.; Hosokawa, H.; et al. Abstract 8: The Membrane-associated Guanylate Kinase Ww and Pdz Domain-containing Protein 1 magi1 is Required for Disturbed Flow-induced Endothelial Inflammation and Atherosclerotic Plaque Formation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, A8. [Google Scholar] [CrossRef]
- Le, N.-T.; Takei, Y.; Izawa-Ishizawa, Y.; Heo, K.-S.; Lee, H.; Smrcka, A.V.; Miller, B.L.; Ko, K.A.; Ture, S.; Morrell, C.; et al. Identification of Activators of ERK5 Transcriptional Activity by High-Throughput Screening and the Role of Endothelial ERK5 in Vasoprotective Effects Induced by Statins and Antimalarial Agents. J. Immunol. 2014, 193, 3803–3815. [Google Scholar] [CrossRef]
- Chen, G.-F.; Sudhahar, V.; Youn, S.-W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T.; et al. Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef] [Green Version]
- Kotla, S.; Vu, H.T.; Ko, K.A.; Wang, Y.; Imanishi, M.; Heo, K.-S.; Fujii, Y.; Thomas, T.N.; Gi, Y.J.; Mazhar, H.; et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2–interacting protein. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Addgene: CRISPR Guide. Available online: https://www.addgene.org/crispr/guide/ (accessed on 27 October 2019).
- Concordet, J.-P.; Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zuñiga, J.; Edison, E.; Palla, S.; Dong, W.; Parker-Thornburg, J. Superovulation Strategies for 6 Commonly Used Mouse Strains. J. Am. Assoc. Lab. Anim. Sci. JAALAS 2011, 50, 471–478. [Google Scholar] [PubMed]
- Kluesner, M.G.; Nedveck, D.A.; Lahr, W.S.; Garbe, J.R.; Abrahante, J.E.; Webber, B.R.; Moriarity, B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2018, 1, 239–250. [Google Scholar] [CrossRef]
- Norton, W.B.; Scavizzi, F.; Smith, C.N.; Dong, W.; Raspa, M.; Parker-Thornburg, J.V. Refinements for embryo implantation surgery in the mouse: comparison of injectable and inhalant anesthesias—Tribromoethanol, ketamine and isoflurane—On pregnancy and pup survival. Lab. Anim. 2016, 50, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Smith, H.E.; Liu, C.; Willi, M.; Hennighausen, L. Cytosine base editor 4 but not adenine base editor generates off-target mutations in mouse embryos. Commun. Biol. 2020, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, X.; He, S.; Huang, S.; Li, C.; Chen, Y.; Liu, Z.; Huang, X.; Wang, X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [Green Version]
- King, R.W.; Jackson, P.K.; Kirschner, M.W. Mitosis in transition. Cell 1994, 79, 563–571. [Google Scholar] [CrossRef]
- Ogura, Y.; Sasakura, Y. Emerging mechanisms regulating mitotic synchrony during animal embryogenesis. Dev. Growth Differ. 2017, 59, 565–579. [Google Scholar] [CrossRef]
- Cooper, G.M. The Eukaryotic Cell Cycle. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Wienert, B.; Shin, J.; Zelin, E.; Pestal, K.; Corn, J.E. In vitro—transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLOS Biol. 2018, 16, e2005840. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell 2015, 160, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Groß, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Zhuang, F.; Hu, X.; Wang, B.; Wen, X.-Z.; Ji, J.-F.; Xi, J.J. Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res. 2017, 27, 578–581. [Google Scholar] [CrossRef] [Green Version]
- Aird, E.J.; Lovendahl, K.N.; Martin, A.S.; Harris, R.S.; Gordon, W.R. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 2018, 1, 54. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.L.; Leroy, P.; Unoson, C.; Fange, D.; Ćurić, V.; Lawson, M.J.; Elf, J. Kinetics of dCas9 target search in Escherichia coli. Science 2017, 357, 1420–1424. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.-C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Project | sgRNA/ Donor Pair | Total | Number Morulae | Number Blasts | Number DNP | Number Dead | % Morulae | % Blasts | % Combined | % DNP | % Dead |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Magi 1 | M67 | 44 | 13 | 11 | 15 | 5 | 30% | 25% | 55% | 34% | 11% |
| M79 | 45 | 6 | 25 | 13 | 1 | 13% | 56% | 69% | 29% | 2% | |
| M79R | 51 | 7 | 15 | 23 | 6 | 14% | 29% | 43% | 45% | 12% | |
| M91 | 41 | 7 | 19 | 13 | 2 | 17% | 46% | 63% | 32% | 5% | |
| Mapk7 | E84 | 48 | 3 | 22 | 19 | 4 | 6% | 46% | 52% | 40% | 8% |
| E87 | 51 | 10 | 23 | 17 | 1 | 20% | 45% | 65% | 33% | 2% | |
| Atox1 | A27 | 22 | 6 | 5 | 10 | 1 | 27% | 23% | 50% | 45% | 5% |
| A43 | 19 | 3 | 6 | 6 | 4 | 16% | 32% | 47% | 32% | 21% |
| Project Designation | Gene (Guide RNA Used) | Mouse Designation | Pups with Desired Mutation/All Pups | Percentage F0 Pups with Desired Mutation | Percentage Blasts with Desired Mutation |
|---|---|---|---|---|---|
| GEMF-748 | mMagi1 (M79) | Magi1-S733A-KI | 5/7 | 71% | 54% |
| GEMF-374 | mMapK7 (E84) | Erk5-S496A-KI | 2/9 | 22% | 39% |
| GEMF-495 | mAtox1 (A27) | Atox1-K3R-KI | 4/20 | 20% | 13% |
| GEMF-1128 | mTerf2ip (T77) | Terf2ip-S202A-KI | 4/18 | 22% | 38% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McBeath, E.; Parker-Thornburg, J.; Fujii, Y.; Aryal, N.; Smith, C.; Hofmann, M.-C.; Abe, J.-i.; Fujiwara, K. Rapid Evaluation of CRISPR Guides and Donors for Engineering Mice. Genes 2020, 11, 628. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060628
McBeath E, Parker-Thornburg J, Fujii Y, Aryal N, Smith C, Hofmann M-C, Abe J-i, Fujiwara K. Rapid Evaluation of CRISPR Guides and Donors for Engineering Mice. Genes. 2020; 11(6):628. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060628
Chicago/Turabian StyleMcBeath, Elena, Jan Parker-Thornburg, Yuka Fujii, Neeraj Aryal, Chad Smith, Marie-Claude Hofmann, Jun-ichi Abe, and Keigi Fujiwara. 2020. "Rapid Evaluation of CRISPR Guides and Donors for Engineering Mice" Genes 11, no. 6: 628. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060628