Gut Microbiota between Environment and Genetic Background in Familial Mediterranean Fever (FMF)

 , , and
, , and 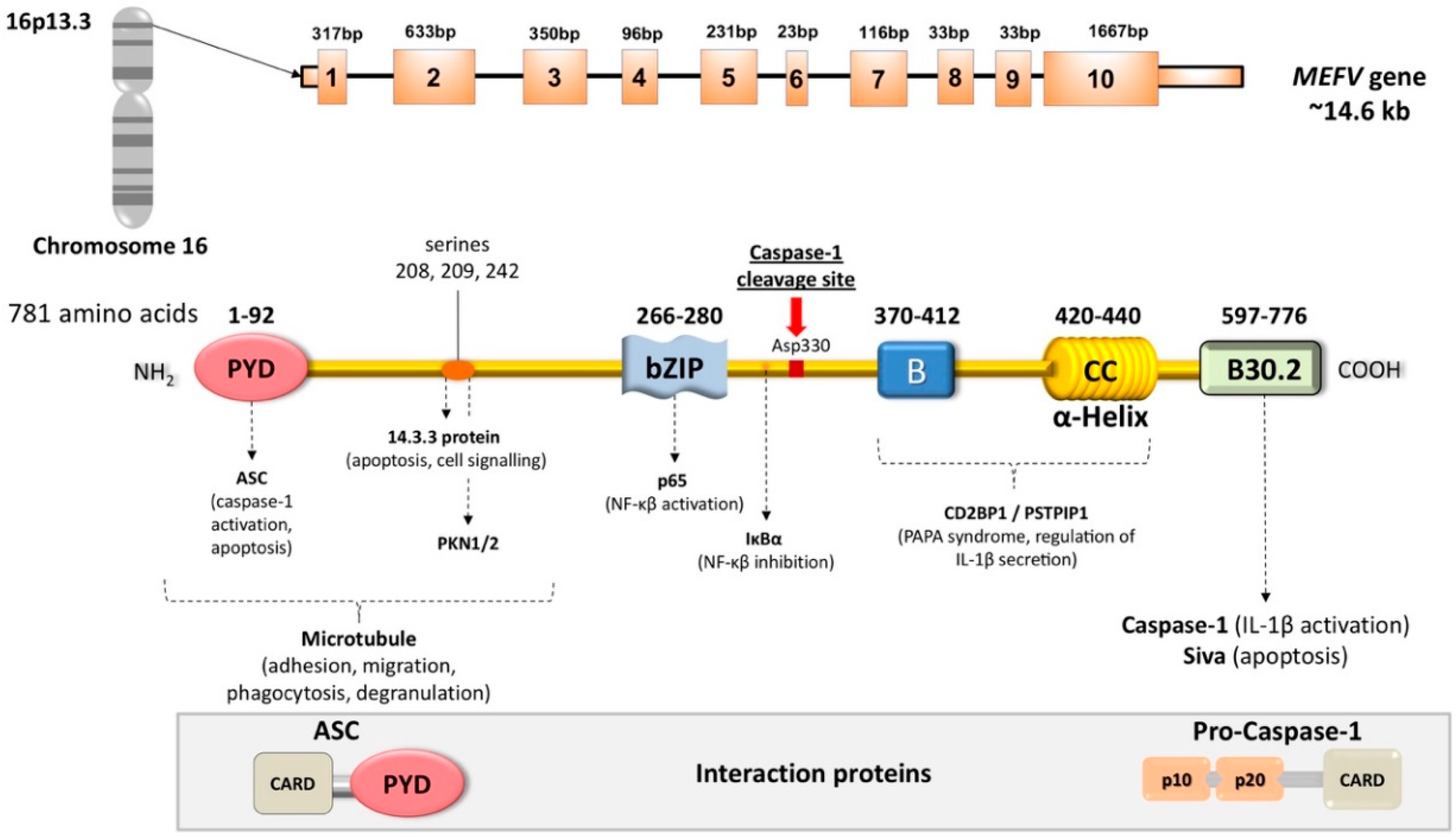{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
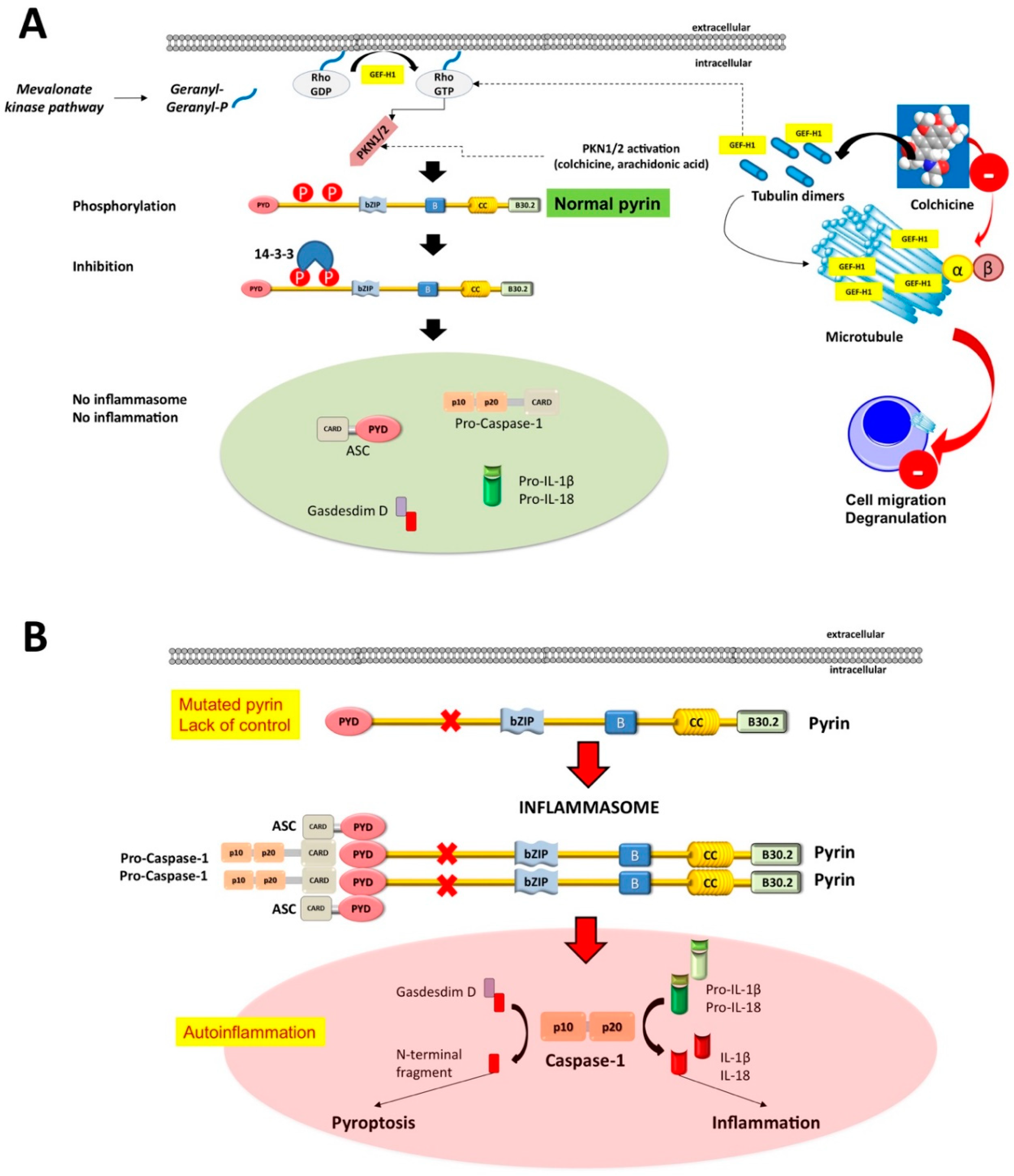
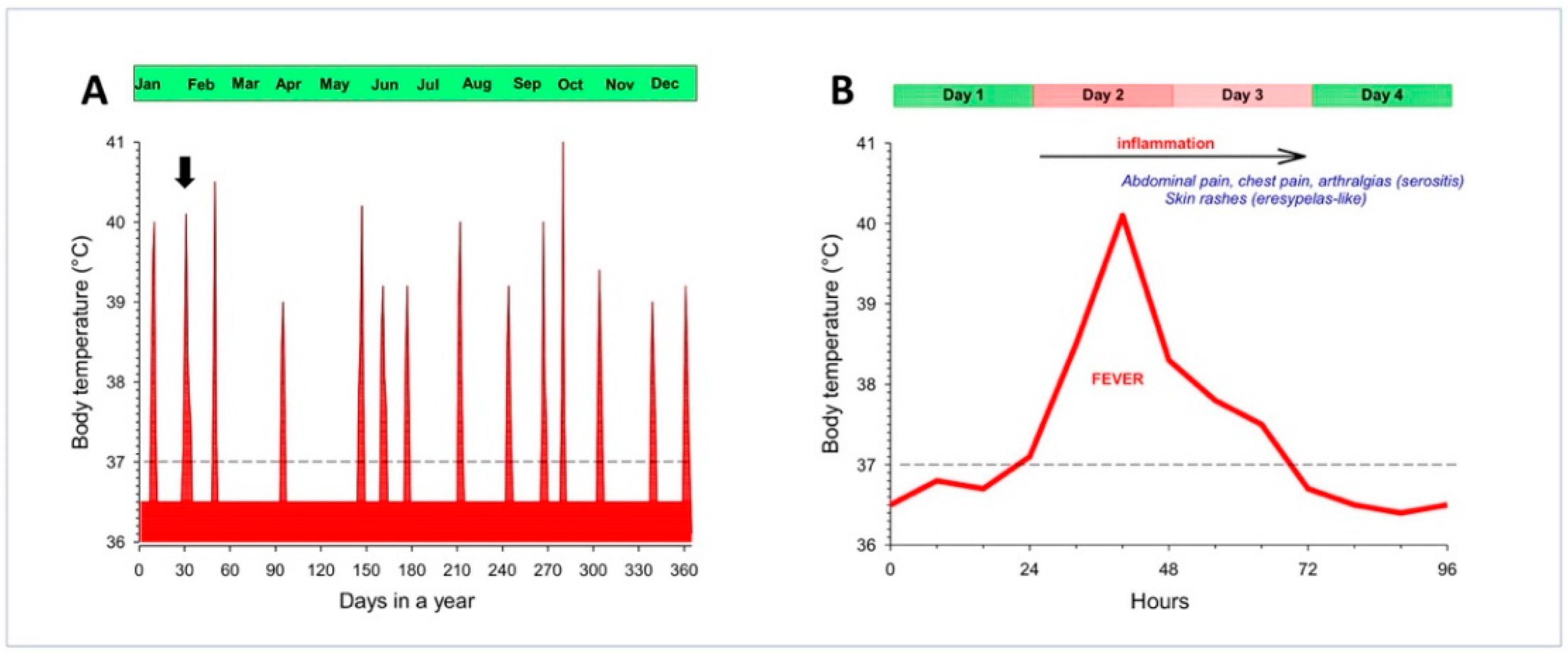
2. Familial Mediterranean Fever
3. Gut Microbiota
3.1. Development of the Gastrointestinal Microbiota
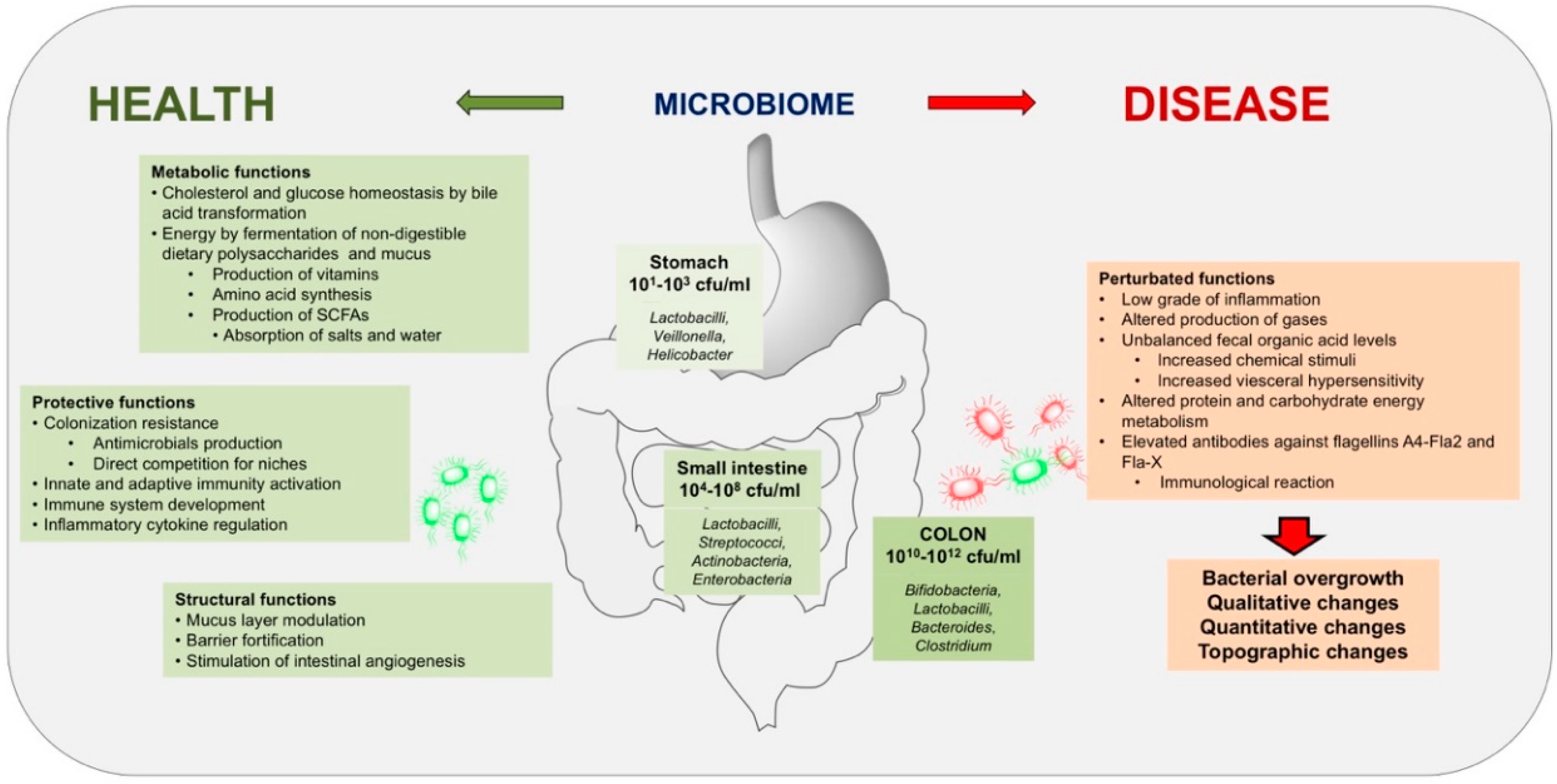
3.2. Characterization of the Gut Microbiota
3.3. Main Features of Bacterial Communities in the Human Intestine
3.4. Functions of the Intestinal Microbiota
3.5. The Microbiota and the Brain-Gut Axis
4. Gene-Bacteria Interplay and Composition of Gut Microbiota in FMF Patients
5. The Role of Environmental Factors and the Determination of Phenotype
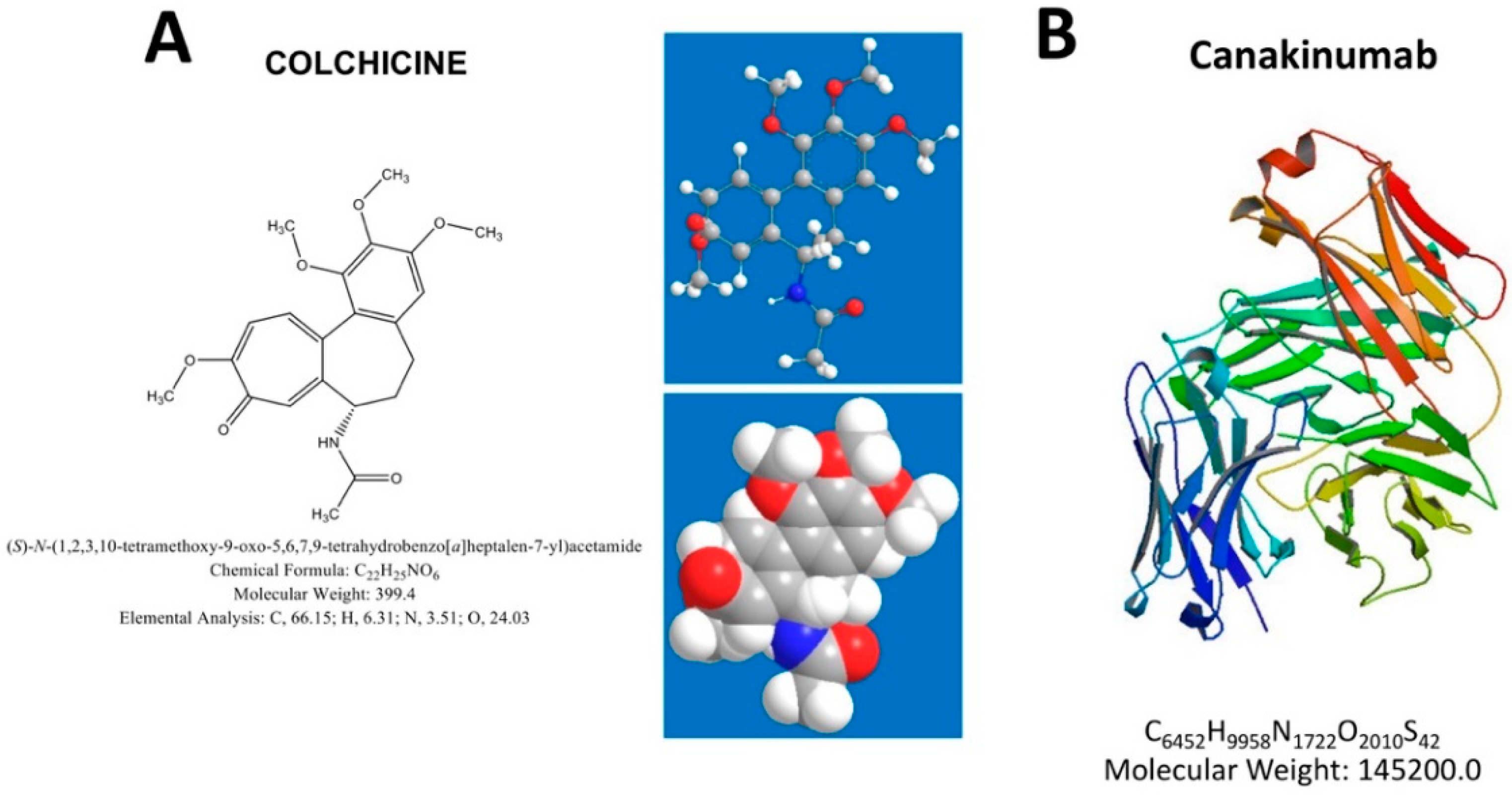
6. Possible Therapeutic Implications
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuramitsu, H.K.; He, X.; Lux, R.; Anderson, M.H.; Shi, W. Interspecies interactions within oral microbial communities. Microbiol. Mol. Biol. Rev. 2007, 71, 653–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfrate, L.; Tack, J.; Grattagliano, I.; Cuomo, R.; Portincasa, P. Microbiota in health and irritable bowel syndrome: Current knowledge, perspectives and therapeutic options. Scand. J. Gastroenterol. 2013, 48, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Smidt, H.; Rijkers, G.T.; de Vos, W.M. Intestinal microbiota in human health and disease: The impact of probiotics. Genes Nutr. 2011, 6, 209–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.B. The intestinal microbiota in health and disease. Curr. Opin. Gastroenterol. 2012, 28, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, W.; Lan, P.; Mou, X. The microbiome in inflammatory bowel diseases: From pathogenesis to therapy. Protein Cell 2020. [Google Scholar] [CrossRef]
- Mari, A.; Abu Baker, F.; Mahamid, M.; Sbeit, W.; Khoury, T. The Evolving Role of Gut Microbiota in the Management of Irritable Bowel Syndrome: An Overview of the Current Knowledge. J. Clin. Med. 2020, 9, 685. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontol 2000 2000, 83, 14–25. [Google Scholar] [CrossRef]
- Pieczynska, M.D.; Yang, Y.; Petrykowski, S.; Horbanczuk, O.K.; Atanasov, A.G.; Horbanczuk, J.O. Gut Microbiota and Its Metabolites in Atherosclerosis Development. Molecules 2020, 25, 594. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castillo, Z.; Valdes-Miramontes, E.; Llamas-Covarrubias, M.; Munoz-Valle, J.F. Troublesome friends within us: The role of gut microbiota on rheumatoid arthritis etiopathogenesis and its clinical and therapeutic relevance. Clin. Exp. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Goodarzi, M.O. Metabolites Linking the Gut Microbiome with Risk for Type 2 Diabetes. Curr. Nutr. Rep. 2020, 9, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Crovesy, L.; Masterson, D.; Rosado, E.L. Profile of the gut microbiota of adults with obesity: A systematic review. Eur. J. Clin. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.C.; Dagher, I.A.; Kloepfer, K.M. Role of the Microbiome in Allergic Disease Development. Curr. Allergy Asthma Rep. 2020, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Abu-Ghazaleh, N.; Chua, W.J.; Gopalan, V. Intestinal microbiota and its association with colon cancer and red/processed meat consumption. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe. 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.A.; Bacigalupe, R.; Wang, J.; Ruhlemann, M.C.; Tito, R.Y.; Falony, G.; Joossens, M.; Vieira-Silva, S.; Henckaerts, L.; Rymenans, L.; et al. Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 2020, 5, 1079–1087. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Akkermans, A.D.L.; Akkermans van-Vliet, W.M.; de Visser, J.A.; de Vos, W.M. The host genotype affects the bacterial community in the human gastrointestinal tract. Microb. Ecol. Health Dis. 2001, 13, 129–134. [Google Scholar] [CrossRef]
- Stewart, J.A.; Chadwick, V.S.; Murray, A. Investigations into the influence of host genetics on the predominant eubacteria in the faecal microflora of children. J. Med. Microbiol. 2005, 54, 1239–1242. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Ozen, S.; Bilginer, Y. A clinical guide to autoinflammatory diseases: Familial Mediterranean fever and next-of-kin. Nat. Rev. Rheumatol. 2014, 10, 135–147. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Touitou, I. Familial mediterranean Fever in the world. Arthritis Rheum. 2009, 61, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Bonfrate, L.; Scaccianoce, G.; Palasciano, G.; Ben-Chetrit, E.; Portincasa, P. A novel cluster of patients with Familial Mediterranean Fever (FMF) in southern Italy. Eur. J. Clin. Investig. 2017, 47, 622–629. [Google Scholar] [CrossRef]
- Ricci, P.; Stella, A.; Settimo, E.; Passerini, F.; Minerva, F.; Belfiore, A.; Palmieri, V.O.; Pugliese, S.; Scaccianoce, G.; Portincasa, P. The grandfather’s fever. Clin. Rheumatol 2019, 39, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Ben-Chetrit, E.; Levy, M. Familial Mediterranean fever. Lancet 1998, 351, 659–664. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Zemer, D.; Zaks, N.; Kees, S.; Lidar, T.; Migdal, A.; Padeh, S.; Pras, M. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997, 40, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Demirkaya, E.; Erer, B.; Livneh, A.; Ben-Chetrit, E.; Giancane, G.; Ozdogan, H.; Abu, I.; Gattorno, M.; Hawkins, P.N.; et al. EULAR recommendations for the management of familial Mediterranean fever. Ann. Rheum. Dis. 2016, 75, 644–651. [Google Scholar] [CrossRef]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E.; et al. Classification criteria for autoinflammatory recurrent fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Van Gijn, M.E.; Ceccherini, I.; Shinar, Y.; Carbo, E.C.; Slofstra, M.; Arostegui, J.I.; Sarrabay, G.; Rowczenio, D.; Omoyimni, E.; Balci-Peynircioglu, B.; et al. New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J. Med. Genet. 2018, 55, 530–537. [Google Scholar] [CrossRef]
- Milhavet, F.; Cuisset, L.; Hoffman, H.M.; Slim, R.; El-Shanti, H.; Aksentijevich, I.; Lesage, S.; Waterham, H.; Wise, C.; Sarrauste de Menthiere, C.; et al. The infevers autoinflammatory mutation online registry: Update with new genes and functions. Hum. Mutat. 2008, 29, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Touitou, I.; Lesage, S.; McDermott, M.; Cuisset, L.; Hoffman, H.; Dode, C.; Shoham, N.; Aganna, E.; Hugot, J.P.; Wise, C.; et al. Infevers: An evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 2004, 24, 194–198. [Google Scholar] [CrossRef] [PubMed]
- De Menthiere, C.S.; Terriere, S.; Pugnere, D.; Ruiz, M.; Demaille, J.; Touitou, I. INFEVERS: The Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res. 2003, 31, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, H.; Huang, L.; Saavedra, P.; Vuylsteke, M.; Asaoka, T.; Prencipe, G.; Insalaco, A.; Ogunjimi, B.; Jeyaratnam, J.; Cataldo, I.; et al. Blood-based test for diagnosis and functional subtyping of familial Mediterranean fever. Ann. Rheum. Dis. 2020, 79, 960–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, A.; Cortellessa, F.; Scaccianoce, G.; Pivetta, B.; Settimo, E.; Portincasa, P. Familial Mediterranean fever: Breaking all the (genetic) rules. Rheumatology 2019, 58, 463–467. [Google Scholar] [CrossRef]
- Accetturo, M.; D’Uggento, A.M.; Portincasa, P.; Stella, A. Improvement of MEFV gene variants classification to aid treatment decision making in familial Mediterranean fever. Rheumatology 2020, 59, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Masters, S.L.; Lagou, V.; Jeru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 2016, 8, 332ra45. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Yang, J.; Liu, W.; Wang, Y.; Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 2016, 113, E4857–E4866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gorp, H.; Saavedra, P.H.; de Vasconcelos, N.M.; Van Opdenbosch, N.; Vande Walle, L.; Matusiak, M.; Prencipe, G.; Insalaco, A.; Van Hauwermeiren, F.; Demon, D.; et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 2016, 113, 14384–14389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Walle, L.V.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Portincasa, P.; Scaccianoce, G.; Palasciano, G. Familial mediterranean fever: A fascinating model of inherited autoinflammatory disorder. Eur. J. Clin. Investig. 2013, 43, 1314–1327. [Google Scholar] [CrossRef]
- Grattagliano, I.; Bonfrate, L.; Ruggiero, V.; Scaccianoce, G.; Palasciano, G.; Portincasa, P. Novel therapeutics for the treatment of familial Mediterranean fever: From colchicine to biologics. Clin. Pharmacol. Ther. 2014, 95, 89–97. [Google Scholar] [CrossRef]
- Portincasa, P. Colchicine, Biologic Agents and More for the Treatment of Familial Mediterranean Fever. The Old, the New, and the Rare. Curr. Med. Chem. 2016, 23, 60–86. [Google Scholar] [CrossRef]
- Wang, D.; Bonfrate, L.; de Bari, O.; Wang, T.; Portincasa, P. Familial Mediterranean Fever: From Pathogenesis to Treatment. J. Genet. Syndr. Gene Ther. 2014, 5, 1000248. [Google Scholar]
- Chae, J.J.; Aksentijevich, I.; Kastner, D.L. Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br. J. Haematol. 2009, 146, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papin, S.; Cuenin, S.; Agostini, L.; Martinon, F.; Werner, S.; Beer, H.D.; Grutter, C.; Grutter, M.; Tschopp, J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007, 14, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Richards, N.; Schaner, P.; Diaz, A.; Stuckey, J.; Shelden, E.; Wadhwa, A.; Gumucio, D.L. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 2001, 276, 39320–39329. [Google Scholar] [CrossRef] [Green Version]
- Shoham, N.G.; Centola, M.; Mansfield, E.; Hull, K.M.; Wood, G.; Wise, C.A.; Kastner, D.L. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 13501–13506. [Google Scholar] [CrossRef] [Green Version]
- Balci-Peynircioglu, B.; Waite, A.L.; Hu, C.; Richards, N.; Staubach-Grosse, A.; Yilmaz, E.; Gumucio, D.L. Pyrin, product of the MEFV locus, interacts with the proapoptotic protein, Siva. J. Cell. Physiol. 2008, 216, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, A.; Malireddi, R.K.S.; Saavedra, P.H.V.; Vande Walle, L.; Van Gorp, H.; Kambara, H.; Tillman, H.; Vogel, P.; Luo, H.R.; Xavier, R.J.; et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J. Exp. Med. 2018, 215, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Van Gorp, H.; Van Opdenbosch, N.; Lamkanfi, M. Inflammasome-Dependent Cytokines at the Crossroads of Health and Autoinflammatory Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a028563. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Chae, J.J.; Wood, G.; Masters, S.L.; Richard, K.; Park, G.; Smith, B.J.; Kastner, D.L. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. USA 2006, 103, 9982–9987. [Google Scholar] [CrossRef] [Green Version]
- Giaglis, S.; Papadopoulos, V.; Kambas, K.; Doumas, M.; Tsironidou, V.; Rafail, S.; Kartalis, G.; Speletas, M.; Ritis, K. MEFV alterations and population genetics analysis in a large cohort of Greek patients with familial Mediterranean fever. Clin. Genet. 2007, 71, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Mattit, H.; Joma, M.; Al-Cheikh, S.; El-Khateeb, M.; Medlej-Hashim, M.; Salem, N.; Delague, V.; Megarbane, A. Familial Mediterranean fever in the Syrian population: Gene mutation frequencies, carrier rates and phenotype-genotype correlation. Eur. J. Med. Genet. 2006, 49, 481–486. [Google Scholar] [CrossRef]
- Goldfinger, S.E. Colchicine for familial Mediterranean fever. N. Engl. J. Med. 1972, 287, 1302. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Wolff, S.M.; Goldfinger, S.E.; Dale, D.C.; Alling, D.W. Colchicine therapy for familial mediterranean fever. A double-blind trial. N. Engl. J. Med. 1974, 291, 934–937. [Google Scholar] [CrossRef]
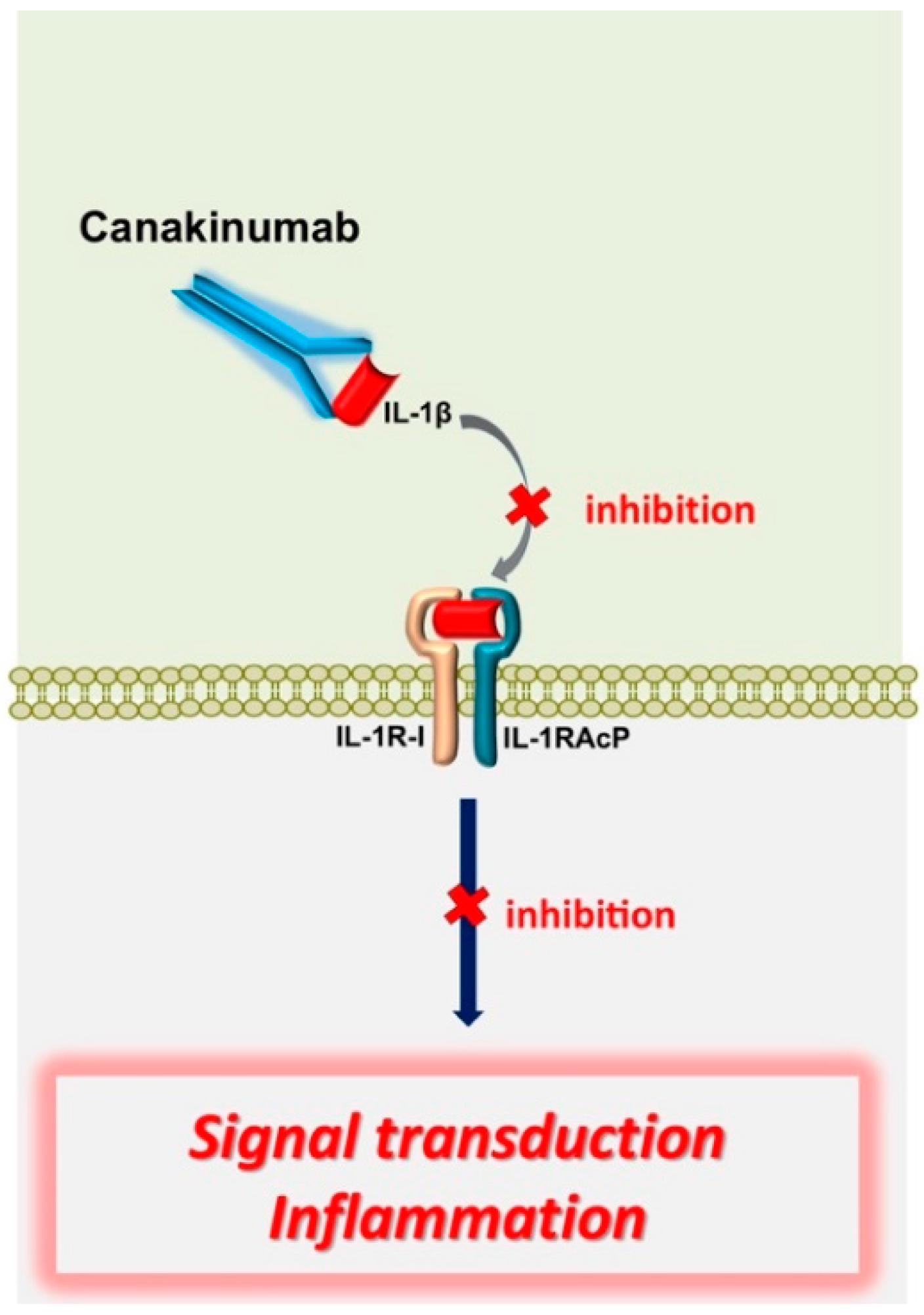
- Gul, A.; Ozdogan, H.; Erer, B.; Ugurlu, S.; Kasapcopur, O.; Davis, N.; Sevgi, S. Efficacy and safety of canakinumab in adolescents and adults with colchicine-resistant familial Mediterranean fever. Arthritis Res. Ther. 2015, 17, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brik, R.; Butbul-Aviel, Y.; Lubin, S.; Dayan, E.B.; Rachmilewitz-Minei, T.; Tseng, L.; Hashkes, P.J. Canakinumab for the treatment of children with colchicine-resistant familial Mediterranean fever: A 6-month open-label, single-arm pilot study. Arthritis Rheumatol. 2014, 66, 3241–3243. [Google Scholar] [CrossRef] [PubMed]
- Van der Hilst, J.; Moutschen, M.; Messiaen, P.E.; Lauwerys, B.R.; Vanderschueren, S. Efficacy of anti-IL-1 treatment in familial Mediterranean fever: A systematic review of the literature. Biol. Targets Ther. 2016, 10, 75–80. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Kone-Paut, I.; Lachmann, H.J.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef] [Green Version]
- Maynard, C.; Weinkove, D. The gut microbiota and ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science; Springer: Berlin/Heidelberg, Germany, 2018; pp. 351–371. [Google Scholar]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.M.; Meyer, K.M.; Prince, A.L.; Aagaard, K.M. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes 2016, 7, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Vandenplas, Y.; Carnielli, V.P.; Ksiazyk, J.; Luna, M.S.; Migacheva, N.; Mosselmans, J.M.; Picaud, J.C.; Possner, M.; Singhal, A.; Wabitsch, M. Factors affecting early-life intestinal microbiota development. Nutrition 2020, 78, 110812. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Sanz, Y. Gut microbiota and probiotics in maternal and infant health. Am. J. Clin. Nutr. 2011, 94, 2000S–2005S. [Google Scholar] [CrossRef]
- Cheng, J.; Palva, A.M.; de Vos, W.M.; Satokari, R. Contribution of the intestinal microbiota to human health: From birth to 100 years of age. In Between Pathogenicity and Commensalism; Springer: Berlin/Heidelberg, Germany, 2011; pp. 323–346. [Google Scholar]
- Bezirtzoglou, E. The intestinal microflora during the first weeks of life. Anaerobe 1997, 3, 173–177. [Google Scholar] [CrossRef]
- Adlerberth, I.; Wold, A. Establishment of the gut microbiota in Western infants. Acta Paediatr. 2009, 98, 229–238. [Google Scholar] [CrossRef]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of faecal Bifidobacterium populations in breast-and formula-fed infants during the first 18 months of life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, L.C.; McCartney, A.L. Longitudinal investigation of the faecal microbiota of healthy full-term infants using fluorescence in situ hybridization and denaturing gradient gel electrophoresis. Microbiology 2010, 156, 3317–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumney, C.J.; Rowland, I.R. In vivo and in vitro models of the human colonic flora. Crit. Rev. Food Sci. Nutr. 1992, 31, 299–331. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajilić-Stojanović, M.; Smidt, H.; De Vos, W.M. Diversity of the human gastrointestinal tract microbiota revisited. Environ. Microbiol. 2007, 9, 2125–2136. [Google Scholar] [CrossRef]
- Egert, M.; De Graaf, A.A.; Maathuis, A.; De Waard, P.; Plugge, C.M.; Smidt, H.; Deutz, N.E.; Dijkema, C.; De Vos, W.M.; Venema, K. Identification of glucose-fermenting bacteria present in an in vitro model of the human intestine by RNA-stable isotope probing. FEMS Microbiol. Ecol. 2007, 60, 126–135. [Google Scholar] [CrossRef]
- Ben-Amor, K.; Heilig, H.; Smidt, H.; Vaughan, E.E.; Abee, T.; de Vos, W.M. Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl. Environ. Microbiol. 2005, 71, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Rinttilä, T.; Kassinen, A.; Malinen, E.; Krogius, L.; Palva, A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl. Microbiol. 2004, 97, 1166–1177. [Google Scholar] [CrossRef]
- Amann, R.; Fuchs, B.M. Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat. Rev. Microbiol. 2008, 6, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Caughey, B. A simplified recipe for prions. Proc. Natl. Acad. Sci. USA 2007, 104, 9551–9552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoetendal, E.; Rajilić-Stojanović, M.; De Vos, W. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008, 57, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogert, B.; de Vos, W.M.; Zoetendal, E.G.; Kleerebezem, M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 2011, 77, 2071–2080. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Jumpstart Reference Strains Consortium. A catalog of reference genomes from the human microbiome. Science 2010, 328, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Booijink, C.C.; Zoetendal, E.G.; Kleerebezem, M.; De Vos, W.M. Microbial communities in the human small intestine: Coupling diversity to metagenomics. Future Microbiol. 2007, 2, 285–295. [Google Scholar] [CrossRef]
- Altomare, D.F.; Bonfrate, L.; Krawczyk, M.; Lammert, F.; Caputi-Jambrenghi, O.; Rizzi, S.; Vacca, M.; Portincasa, P. The inulin hydrogen breath test predicts the quality of colonic preparation. Surg. Endosc. 2014, 28, 1579–1587. [Google Scholar] [CrossRef]
- Bonfrate, L.; Di Palo, D.M.; Celano, G.; Albert, A.; Vitellio, P.; De Angelis, M.; Gobbetti, M.; Portincasa, P. Effects of Bifidobacterium longum BB536 and Lactobacillus rhamnosus HN001 in IBS patients. Eur. J. Clin. Investig. 2020, 50, e13201. [Google Scholar] [CrossRef]
- De Angelis, M.; Garruti, G.; Minervini, F.; Bonfrate, L.; Portincasa, P.; Gobbetti, M. The Food-gut Human Axis: The Effects of Diet on Gut Microbiota and Metabolome. Curr. Med. Chem. 2019, 26, 3567–3583. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, 4–14. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; de Bari, O.; Lembo, A.; Ballou, S. Irritable bowel syndrome and diet. Gastroenterol. Rep. 2017, 5, 11–19. [Google Scholar] [CrossRef]
- Quigley, E.M. Microflora modulation of motility. J. Neurogastroenterol. Motil. 2011, 17, 140. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Mans, J.J.; von Lackum, K.; Dorsey, C.; Willis, S.; Wallet, S.M.; Baker, H.V.; Lamont, R.J.; Handfield, M. The degree of microbiome complexity influences the epithelial response to infection. BMC Genom. 2009, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [Green Version]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-Talk Between Bile Acids and Gastro-Intestinal and Thermogenic Hormones: Clues from Bariatric Surgery. Ann. Hepatol. 2017, 16, S68–S82. [Google Scholar] [CrossRef]
- Garruti, G.; Wang, D.Q.; Di Ciaula, A.; Portincasa, P. Cholecystectomy: A way forward and back to metabolic syndrome? Lab. Invest. 2018, 98, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Hijova, E.; Chmelarova, A. Short chain fatty acids and colonic health. Bratisl. Med. J. 2007, 108, 354–358. [Google Scholar]
- Chen, W.-J.L.; Anderson, J.W.; Jennings, D. Propionate may mediate the hypocholesterolemic effects of certain soluble plant fibers in cholesterol-fed rats. Exp. Biol. Med. 1984, 175, 215–218. [Google Scholar] [CrossRef]
- Machado, M.V.; Cortez-Pinto, H. Gut microbiota and nonalcoholic fatty liver disease. Ann. Hepatol. 2012, 11, 440–449. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [Green Version]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Lawson, M.A.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Prakash, S.; Rodes, L.; Coussa-Charley, M.; Tomaro-Duchesneau, C. Gut microbiota: Next frontier in understanding human health and development of biotherapeutics. Biol. Targets Ther. 2011, 5, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Lührs, H.; Gerke, T.; Müller, J.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate inhibits NF-κB activation in lamina propria macrophages of patients with ulcerative colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Kleessen, B.; Blaut, M. Modulation of gut mucosal biofilms. Br. J. Nutr. 2005, 93, S35–S40. [Google Scholar] [CrossRef] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.; Brummer, R.J. The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef]
- Drossman, D.A. Gastrointestinal illness and the biopsychosocial model. Psychosom. Med. 1998, 60, 258–267. [Google Scholar] [CrossRef]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain–gut–enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, P.; Kunze, W.A. Voices from within: Gut microbes and the CNS. Cell. Mol. Life Sci. 2013, 70, 55–69. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Dinan, T.G.; Cryan, J.F. The gut microbiota as a key regulator of visceral pain. Pain 2017, 158, S19–S28. [Google Scholar] [CrossRef] [Green Version]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, K.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255-e119. [Google Scholar] [CrossRef]
- Gareau, M.G.; Wine, E.; Rodrigues, D.M.; Cho, J.H.; Whary, M.T.; Philpott, D.J.; MacQueen, G.; Sherman, P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2011, 60, 307–317. [Google Scholar] [CrossRef]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A. Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 2010, 139, 2102–2112. [Google Scholar] [CrossRef] [Green Version]
- Bercik, P.; Park, A.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.; Fahnestock, M.; Moine, D. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut–brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, P.; Kunze, W.A.; Bienenstock, J. On communication between gut microbes and the brain. Curr. Opin. Gastroenterol. 2012, 28, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Quigley, E.M.; Ahmed, S.M.; Scully, P.; O’Brien, S.; O’Mahony, L.; O’Mahony, S.; Shanahan, F.; Keeling, P.N. Hypothalamic-pituitary-gut axis dysregulation in irritable bowel syndrome: Plasma cytokines as a potential biomarker? Gastroenterology 2006, 130, 304–311. [Google Scholar] [CrossRef]
- Goehler, L.E.; Gaykema, R.P.; Opitz, N.; Reddaway, R.; Badr, N.; Lyte, M. Activation in vagal afferents and central autonomic pathways: Early responses to intestinal infection with Campylobacter jejuni. Brain Behav. Immun. 2005, 19, 334–344. [Google Scholar] [CrossRef]
- Hugot, J.-P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.-P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Núñez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Berkun, Y.; Karban, A.; Padeh, S.; Pras, E.; Shinar, Y.; Lidar, M.; Livneh, A.; Bujanover, Y. NOD2/CARD15 gene mutations in patients with familial Mediterranean fever. Semin. Arthritis Rheum. 2012, 42, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Egritas, O.; Dalgic, B. Infantile colitis as a novel presentation of familial Mediterranean fever responding to colchicine therapy. J. Paediatr. Gastroenterol. Nutr. 2011, 53, 102–105. [Google Scholar] [CrossRef]
- Roldan, R.; Ruiz, A.M.; Miranda, M.D.; Collantes, E. Anakinra: New therapeutic approach in children with Familial Mediterranean Fever resistant to colchicine. Jt. Bone Spine 2008, 75, 504–505. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Sengul, B.; Yavuzsen, T.U.; Booth, D.R.; Booth, S.E.; Bybee, A.; Gallimore, J.R.; Soyturk, M.; Akar, S.; Tunca, M.; et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology 2006, 45, 746–750. [Google Scholar] [CrossRef] [Green Version]
- Ozen, S.; Kone-Paut, I.; Gül, A. Colchicine resistance and intolerance in familial mediterranean fever: Definition, causes, and alternative treatments. Semin. Arthritis Rheum. 2017, 47, 115–120. [Google Scholar] [CrossRef]
- Cattan, D. MEFV mutation carriers and diseases other than familial Mediterranean fever: Proved and non-proved associations; putative biological advantage. Curr. Drug Targets. Inflamm. Allergy 2005, 4, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Cerquaglia, C.; Diaco, M.; Nucera, G.; La Regina, M.; Montalto, M.; Manna, R. Pharmacological and clinical basis of treatment of Familial Mediterranean Fever (FMF) with colchicine or analogues: An update. Curr. Drug Targets. Inflamm. Allergy 2005, 4, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Stella, A.; Bagnulo, R.; Ruggiero, V.; Bonfrate, L.; Castorani, L.; Scaccianoce, G.; Resta, N.; Palasciano, G. Genotypical/phenotypical characterization of a novel cluster of patients with familial Mediterranean fever (FMF) in Apulia and Lucania in Southern Italy. Eur. J. Clin. Investig. 2013, 43, 72. [Google Scholar]
- Chae, J.J.; Komarow, H.D.; Cheng, J.; Wood, G.; Raben, N.; Liu, P.P.; Kastner, D.L. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 2003, 11, 591–604. [Google Scholar] [CrossRef]
- Demirturk, L.; Ozel, A.M.; Cekem, K.; Yazgan, Y.; Gultepe, M. Co-existence of Helicobacter pylori infection in patients with Familial Mediterranean Fever (FMF) and the effect of Helicobacter pylori on the frequency and severity of FMF attacks. Dig. Liver Dis. 2005, 37, 153–158. [Google Scholar] [CrossRef]
- Ozel, A.M.; Demirturk, L.; Aydogdu, A.; Gultepe, M.; Yazgan, Y.; Imirzalioglu, N.; Gurbuz, A.K.; Narin, Y. Effect of Helicobacter pylori infection and eradication therapy on interleukin-6 levels in patients with Familial Mediterranean Fever. Int. J. Clin. Pract. 2008, 62, 754–761. [Google Scholar] [CrossRef]
- Ghoshal, U.C.; Shukla, R.; Ghoshal, U. Small Intestinal Bacterial Overgrowth and Irritable Bowel Syndrome: A Bridge between Functional Organic Dichotomy. Gut Liver 2017, 11, 196–208. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Gerardi, V.; Gasbarrini, A. Diagnosis and treatment of small intestinal bacterial overgrowth. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 215–227. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Corazza, G.R.; Gasbarrini, G.; Montalto, M.; Di Stefano, M.; Basilisco, G.; Parodi, A.; Usai-Satta, P.; Vernia, P.; Anania, C.; et al. Methodology and indications of H2-breath testing in gastrointestinal diseases: The Rome Consensus Conference. Aliment. Pharmacol. Ther. 2009, 29 (Suppl. S1), 1–49. [Google Scholar] [CrossRef]
- Balzan, S.; de Almeida Quadros, C.; de Cleva, R.; Zilberstein, B.; Cecconello, I. Bacterial translocation: Overview of mechanisms and clinical impact. J. Gastroenterol. Hepatol. 2007, 22, 464–471. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Small intestinal bacterial overgrowth: What it is and what it is not. Curr. Opin. Gastroenterol. 2014, 30, 141–146. [Google Scholar] [CrossRef]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 2020, 12, 564. [Google Scholar] [CrossRef] [Green Version]
- Scarpignato, C.; Gatta, L. Commentary: Towards an effective and safe treatment of small intestine bacterial overgrowth. Aliment. Pharmacol. Ther. 2013, 38, 1409–1410. [Google Scholar] [CrossRef]
- Verrecchia, E.; Sicignano, L.L.; La Regina, M.; Nucera, G.; Patisso, I.; Cerrito, L.; Montalto, M.; Gasbarrini, A.; Manna, R. Small Intestinal Bacterial Overgrowth Affects the Responsiveness to Colchicine in Familial Mediterranean Fever. Mediat. Inflamm. 2017, 2017, 7461426. [Google Scholar] [CrossRef]
- Deshayes, S.; Fellahi, S.; Bastard, J.P.; Launay, J.M.; Callebert, J.; Fraisse, T.; Buob, D.; Boffa, J.J.; Giurgea, I.; Dupont, C.; et al. Specific changes in faecal microbiota are associated with familial Mediterranean fever. Ann. Rheum. Dis. 2019, 78, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Khachatryan, Z.A.; Ktsoyan, Z.A.; Manukyan, G.P.; Kelly, D.; Ghazaryan, K.A.; Aminov, R.I. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE 2008, 3, e3064. [Google Scholar] [CrossRef] [Green Version]
- Ktsoyan, Z.A.; Beloborodova, N.V.; Sedrakyan, A.M.; Osipov, G.A.; Khachatryan, Z.A.; Kelly, D.; Manukyan, G.P.; Arakelova, K.A.; Hovhannisyan, A.I.; Olenin, A.Y.; et al. Profiles of Microbial Fatty Acids in the Human Metabolome are Disease-Specific. Front. Microbiol. 2010, 1, 148. [Google Scholar] [CrossRef] [Green Version]
- Ktsoyan, Z.A.; Mkrtchyan, M.S.; Zakharyan, M.K.; Mnatsakanyan, A.A.; Arakelova, K.A.; Gevorgyan, Z.U.; Sedrakyan, A.M.; Hovhannisyan, A.I.; Arakelyan, A.A.; Aminov, R.I. Systemic Concentrations of Short Chain Fatty Acids Are Elevated in Salmonellosis and Exacerbation of Familial Mediterranean Fever. Front. Microbiol. 2016, 7, 776. [Google Scholar] [CrossRef]
- Pepoyan, A.; Balayan, M.; Manvelyan, A.; Galstyan, L.; Pepoyan, S.; Petrosyan, S.; Tsaturyan, V.; Kamiya, S.; Torok, T.; Chikindas, M. Probiotic Lactobacillus acidophilus Strain INMIA 9602 Er 317/402 Administration Reduces the Numbers of Candida albicans and Abundance of Enterobacteria in the Gut Microbiota of Familial Mediterranean Fever Patients. Front. Immunol. 2018, 9, 1426. [Google Scholar] [CrossRef] [Green Version]
- Manukyan, G.P.; Ghazaryan, K.A.; Ktsoyan, Z.A.; Khachatryan, Z.A.; Arakelova, K.A.; Kelly, D.; Grant, G.; Aminov, R.I. Elevated systemic antibodies towards commensal gut microbiota in autoinflammatory condition. PLoS ONE 2008, 3, e3172. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Ktsoyan, Z.A.; Beloborodova, N.V.; Sedrakyan, A.M.; Osipov, G.A.; Khachatryan, Z.A.; Manukyan, G.P.; Arakelova, K.A.; Hovhannisyan, A.I.; Arakelyan, A.A.; Ghazaryan, K.A.; et al. Management of familial Mediterranean fever by colchicine does not normalize the altered profile of microbial long chain fatty acids in the human metabolome. Front. Cell. Infect. Microbiol. 2013, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Posautz, A.; Loncaric, I.; Westermark, P. Is there a connection between the microbiome and AA amyloidosis? First hints from the European brown hare (Lepus europaeus). Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2019, 26, 119–120. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, L.; Chen, S.; Zhou, H.; Fan, Y.; Lin, L.; Li, J.; Xu, J.; Chen, Y.; Ma, Y.; et al. Gut Microbiome Alterations Precede Cerebral Amyloidosis and Microglial Pathology in a Mouse Model of Alzheimer’s Disease. Biomed. Res. Int. 2020, 2020, 8456596. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef]
- Alimov, I.; Menon, S.; Cochran, N.; Maher, R.; Wang, Q.; Alford, J.; Concannon, J.B.; Yang, Z.; Harrington, E.; Llamas, L.; et al. Bile acid analogues are activators of pyrin inflammasome. J. Biol. Chem. 2019, 294, 3359–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theriot, C.M.; Petri, W.A., Jr. Role of Microbiota-Derived Bile Acids in Enteric Infections. Cell 2020, 181, 1452–1454. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, Y.; Huang, J.; Yu, D.; Li, X.; Hu, H.; Ge, T.; Li, D.; et al. Gut Microbiota Dysbiosis Is Associated with Altered Bile Acid Metabolism in Infantile Cholestasis. mSystems 2019, 4, e00463-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michonneau, D.; Latis, E.; Curis, E.; Dubouchet, L.; Ramamoorthy, S.; Ingram, B.; de Latour, R.P.; Robin, M.; de Fontbrune, F.S.; Chevret, S.; et al. Metabolomics analysis of human acute graft-versus-host disease reveals changes in host and microbiota-derived metabolites. Nat. Commun. 2019, 10, 5695. [Google Scholar] [CrossRef] [Green Version]
- Touitou, I.; Sarkisian, T.; Medlej-Hashim, M.; Tunca, M.; Livneh, A.; Cattan, D.; Yalcinkaya, F.; Ozen, S.; Majeed, H.; Ozdogan, H.; et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007, 56, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Aktay, N.; Lainka, E.; Duzova, A.; Bakkaloglu, A.; Kallinich, T. Disease severity in children and adolescents with familial Mediterranean fever: A comparative study to explore environmental effects on a monogenic disease. Ann. Rheum. Dis. 2009, 68, 246–248. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balayan, M.; Manvelyan, A.; Marutyan, S.; Isajanyan, M.; Tsaturyan, V.; Pepoyan, A.; Marotta, F.; Torok, T. Impact of Lactobacillus Acidophilus INMIA 9602 Er-2 and Escherichia coli M-17 on some clinical blood characteristics of familial mediterranean fever disease patients from the armenian cohort. Int. J. Probiotics Prebiotics 2015, 10, 91–95. [Google Scholar]
- Pepoian, A.Z.; Arutunian, N.; Grigorian, A.; Tsaturian, V.V.; Manvelian, A.M.; Dilnian, E.; Balaian, M.A.; Torok, T. The Certain Clinical Characteristics of Blood in Patients with Family Mediterranean Fever of Armenian Population. Klin. Lab. Diagn. 2015, 60, 46–47. [Google Scholar] [PubMed]
- Goulet, O. Potential role of the intestinal microbiota in programming health and disease. Nutr. Rev. 2015, 73 (Suppl. S1), 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yerzinkyan, L.A. Biological Properties of Some Isolates of Lactic Acid Bacteria. Yerevan Acad. Sci. Armen. 1971. [Google Scholar]
- Mkrtchyan, H.; Gibbons, S.; Heidelberger, S.; Zloh, M.; Limaki, H.K. Purification, characterisation and identification of acidocin LCHV, an antimicrobial peptide produced by Lactobacillus acidophilus nv Er 317/402 strain Narine. Int. J. Antimicrob. Agents 2010, 35, 255–260. [Google Scholar] [CrossRef]
- Pepoyan, A.Z.; Balayan, M.H.; Manvelyan, A.M.; Mamikonyan, V.; Isajanyan, M.; Tsaturyan, V.V.; Kamiya, S.; Netrebov, V.; Chikindas, M.L. Lactobacillus acidophilus INMIA 9602 Er-2 strain 317/402 probiotic regulates growth of commensal Escherichia coli in gut microbiota of familial Mediterranean fever disease subjects. Lett. Appl. Microbiol. 2017, 64, 254–260. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Ciaula, A.; Stella, A.; Bonfrate, L.; Wang, D.Q.H.; Portincasa, P. Gut Microbiota between Environment and Genetic Background in Familial Mediterranean Fever (FMF). Genes 2020, 11, 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091041
Di Ciaula A, Stella A, Bonfrate L, Wang DQH, Portincasa P. Gut Microbiota between Environment and Genetic Background in Familial Mediterranean Fever (FMF). Genes. 2020; 11(9):1041. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091041
Chicago/Turabian StyleDi Ciaula, Agostino, Alessandro Stella, Leonilde Bonfrate, David Q. H. Wang, and Piero Portincasa. 2020. "Gut Microbiota between Environment and Genetic Background in Familial Mediterranean Fever (FMF)" Genes 11, no. 9: 1041. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091041