Approaching Shared Pathophysiology in Immune-Mediated Diseases through Functional Genomics

 , and
, and 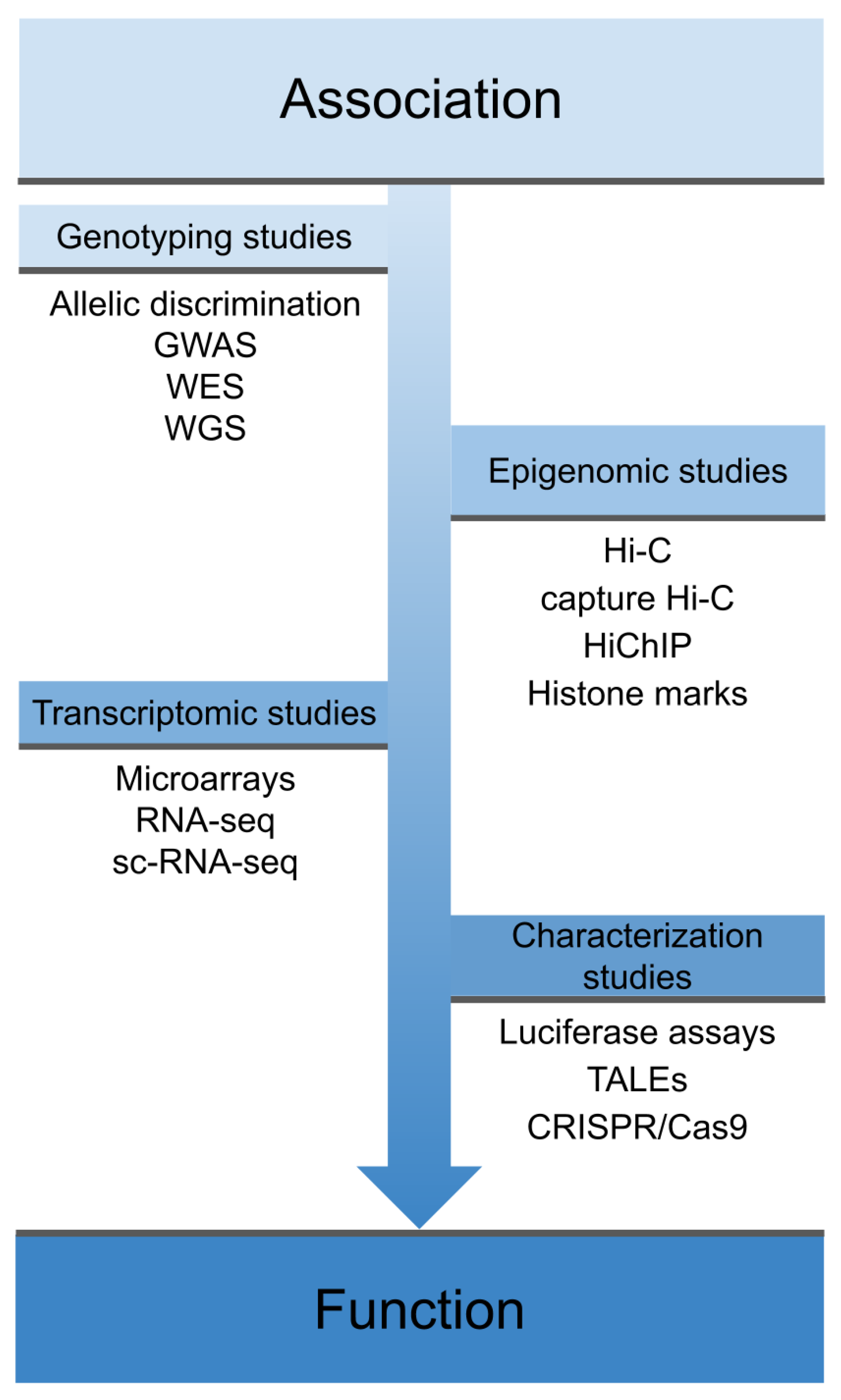{kind=link}
Abstract
:1. Introduction
2. Shared Genetics across IMDs
3. Transcriptomic Studies
4. Epigenomics Studies
5. Experimental Characterization
6. New Approaches for Drug Targeting
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, J.H.; Feldman, M. Heterogeneity of autoimmune diseases: Pathophysiologic insights from genetics and implications for new therapies. Nat. Med. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Mejías, R.; Castañeda, S.; González-Juanatey, C.; Corrales, A.; Ferraz-Amaro, I.; Genre, F.; Remuzgo-Martínez, S.; Rodriguez-Rodriguez, L.; Blanco, R.; Llorca, J.; et al. Cardiovascular risk assessment in patients with rheumatoid arthritis: The relevance of clinical, genetic and serological markers. Autoimmun. Rev. 2016, 15, 1013–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Lipsky, P.E. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2020, 16, 565–579. [Google Scholar] [CrossRef]
- Bournia, V.-K.; Vlachoyiannopoulos, P.G. Subgroups of Sjögren syndrome patients according to serological profiles. J. Autoimmun. 2012, 39, 15–26. [Google Scholar] [CrossRef]
- Parkes, M.; Cortes, A.; van Heel, D.A.; Brown, M.A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013, 14, 661–673. [Google Scholar] [CrossRef]
- Polychronakos, C. Fine points in mapping autoimmunity. Nat. Genet. 2011, 43, 1173–1174. [Google Scholar] [CrossRef]
- Cortes, A.; Brown, M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011, 13, 101. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Bentham, J.; Morris, D.L.; Cunninghame Graham, D.S.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Carmona, F.D.; Vaglio, A.; Mackie, S.L.; Hernández-Rodríguez, J.; Monach, P.A.; Castañeda, S.; Solans, R.; Morado, I.C.; Narváez, J.; Ramentol-Sintas, M.; et al. A genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am. J. Hum. Genet. 2017, 100, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Isac, E.; Acosta-Herrera, M.; Kerick, M.; Assassi, S.; Satpathy, A.T.; Granja, J.; Mumbach, M.R.; Beretta, L.; Simeón, C.P.; Carreira, P.; et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019, 10, 4955. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Frantzeskos, A.; Orozco, G. Functional genomics in autoimmune diseases. Hum. Mol. Genet. 2020, 29, R59–R65. [Google Scholar] [CrossRef]
- Zeggini, E.; Gloyn, A.L.; Barton, A.C.; Wain, L.V. Translational genomics and precision medicine: Moving from the lab to the clinic. Science 2019, 365, 1409–1413. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.-E.; Assassi, S.; Diaz-Gallo, L.-M.; Broen, J.C.; Simeon, C.P.; Castellvi, I.; Vicente-Rabaneda, E.; Fonollosa, V.; Ortego-Centeno, N.; González-Gay, M.A.; et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum. Mol. Genet. 2013, 22, 4021–4029. [Google Scholar] [CrossRef] [Green Version]
- López-Isac, E.; Martín, J.-E.; Assassi, S.; Simeón, C.P.; Carreira, P.; Ortego-Centeno, N.; Freire, M.; Beltrán, E.; Narváez, J.; Alegre-Sancho, J.J.; et al. Brief report: IRF4 newly identified as a common susceptibility locus for systemic sclerosis and rheumatoid arthritis in a cross-disease meta-analysis of genome-wide association studies. Arthritis Rheumatol. 2016, 68, 2338–2344. [Google Scholar] [CrossRef] [Green Version]
- Márquez, A.; Vidal-Bralo, L.; Rodríguez-Rodríguez, L.; González-Gay, M.A.; Balsa, A.; González-Álvaro, I.; Carreira, P.; Ortego-Centeno, N.; Ayala-Gutiérrez, M.M.; García-Hernández, F.J.; et al. A combined large-scale meta-analysis identifies COG6 as a novel shared risk locus for rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.R.; Li, J.; Zhao, S.D.; Bradfield, J.P.; Mentch, F.D.; Maggadottir, S.M.; Hou, C.; Abrams, D.J.; Chang, D.; Gao, F.; et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 2015, 21, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hübenthal, M.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Márquez, A.; Kerick, M.; Zhernakova, A.; Gutierrez-Achury, J.; Chen, W.-M.; Onengut-Gumuscu, S.; González-Álvaro, I.; Rodriguez-Rodriguez, L.; Rios-Fernández, R.; González-Gay, M.A.; et al. Meta-analysis of immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Fernández, L.; Carmona, F.D.; López-Mejías, R.; González-Escribano, M.F.; Lyons, P.A.; Morgan, A.W.; Sawalha, A.H.; Merkel, P.A.; Smith, K.G.C.; González-Gay, M.A.; et al. Cross-phenotype analysis of immunochip data identifies KDM4C as a relevant locus for the development of systemic vasculitis. Ann. Rheum. Dis. 2018, 77, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Herrera, M.; Kerick, M.; González-Serna, D.; Wijmenga, C.; Franke, A.; Gregersen, P.K.; Padyukov, L.; Worthington, J.; Vyse, T.J.; Alarcón-Riquelme, M.E.; et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann. Rheum. Dis. 2019, 78, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, S.; Chen, J.; Xie, X.; Gao, S.; Zhang, C.; Zhou, S.; Wang, J.; Mai, R.; Lin, Q.; et al. Germline genetic patterns underlying familial rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome highlight T cell-initiated autoimmunity. Ann. Rheum. Dis. 2020, 79, 268–275. [Google Scholar] [CrossRef]
- Barturen, G.; Beretta, L.; Cervera, R.; Van Vollenhoven, R.; Alarcón-Riquelme, M.E. Moving towards a molecular taxonomy of autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 180. [Google Scholar] [CrossRef] [Green Version]
- Barturen, G.; Babaei, S.; Català-Moll, F.; Martínez-Bueno, M.; Makowska, Z.; Martorell-Marugán, J.; Carmona-Sáez, P.; Toro-Domínguez, D.; Carnero-Montero, E.; Teruel, M.; et al. Integrative Analysis Reveals a Molecular Stratification of Systemic Autoimmune Diseases. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Y.-F.; Liu, L.; Bielowka, A.; Ahmed, R.; Zhang, H.; Tombleson, P.; Roberts, A.L.; Odhams, C.A.; Cunninghame Graham, D.S.; et al. Genome-wide assessment of genetic risk for systemic lupus erythematosus and disease severity. Hum. Mol. Genet. 2020, 29, 1745–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.A.; Wegmann, D.; Trynka, G.; Gutierrez-Achury, J.; Do, R.; Voight, B.F.; Kraft, P.; Chen, R.; Kallberg, H.J.; Kurreeman, F.A.S.; et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet. 2012, 44, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Bossini-Castillo, L.; Villanueva-Martin, G.; Kerick, M.; Acosta-Herrera, M.; López-Isac, E.; Simeón, C.P.; Ortego-Centeno, N.; Assassi, S.; International SSc Group; Australian Scleroderma Interest Group (ASIG); et al. Genomic risk score impact on susceptibility to systemic sclerosis. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Patrick, M.T.; Stuart, P.E.; Raja, K.; Gudjonsson, J.E.; Tejasvi, T.; Yang, J.; Chandran, V.; Das, S.; Callis-Duffin, K.; Ellinghaus, E.; et al. Genetic signature to provide robust risk assessment of psoriatic arthritis development in psoriasis patients. Nat. Commun. 2018, 9, 1354. [Google Scholar] [CrossRef]
- Knevel, R.; le Cessie, S.; Terao, C.C.; Slowikowski, K.; Cui, J.; Huizinga, T.W.J.; Costenbader, K.H.; Liao, K.P.; Karlson, E.W.; Raychaudhuri, S. Using genetics to prioritize diagnoses for rheumatology outpatients with inflammatory arthritis. Sci. Transl. Med. 2020, 12, eaay1548. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Banchereau, R.; Cepika, A.-M.; Banchereau, J.; Pascual, V. Understanding human autoimmunity and autoinflammation through transcriptomics. Annu. Rev. Immunol. 2017, 35, 337–370. [Google Scholar] [CrossRef] [Green Version]
- Ricaño-Ponce, I.; Zhernakova, D.V.; Deelen, P.; Luo, O.; Li, X.; Isaacs, A.; Karjalainen, J.; Di Tommaso, J.; Borek, Z.A.; Zorro, M.M.; et al. Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non-coding RNAs. J. Autoimmun. 2016, 68, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Ono, Y.; Uno, K.D.; Yamada, R.G.; Ueda, H.R.; Saitou, M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006, 34, e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Saitou, M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat. Protoc. 2007, 2, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA sequencing in cancer: Lessons learned and emerging challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef]
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: Do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.; Korsunsky, I.; Marshall, J.L.; Gao, A.; Watts, G.F.M.; Major, T.; Croft, A.P.; Watts, J.; Blazar, P.E.; Lange, J.K.; et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 2020, 582, 259–264. [Google Scholar] [CrossRef]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Trejo Bittar, H.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Orvain, C.; Assassi, S.; Avouac, J.; Allanore, Y. Systemic sclerosis pathogenesis: Contribution of recent advances in genetics. Curr. Opin. Rheumatol. 2020, 32, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawel, D.R.; Serra-Musach, J.; Lilja, S.; Aagesen, J.; Arenas, A.; Asking, B.; Bengnér, M.; Björkander, J.; Biggs, S.; Ernerudh, J.; et al. A validated single-cell-based strategy to identify diagnostic and therapeutic targets in complex diseases. Genome Med. 2019, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Stavnezer, J.; Schrader, C.E. IgH chain class switch recombination: Mechanism and regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef] [Green Version]
- Bashford-Rogers, R.J.M.; Bergamaschi, L.; McKinney, E.F.; Pombal, D.C.; Mescia, F.; Lee, J.C.; Thomas, D.C.; Flint, S.M.; Kellam, P.; Jayne, D.R.W.; et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 2019, 574, 122–126. [Google Scholar] [CrossRef]
- Singh, M.; Al-Eryani, G.; Carswell, S.; Ferguson, J.M.; Blackburn, J.; Barton, K.; Roden, D.; Luciani, F.; Phan, T.G.; Junankar, S.; et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Van Eerden, S.L.; Standley, D.M.; Teraguchi, S.; Saputri, D.S.; Llamas-Covarrubias, M.A.; Davila, A.; Diez, D.; Nazlica, S.A.; Rozewicki, J.; Ismanto, H.S.; et al. Methods for sequence and structural analysis of B and T cell receptor repertoires. Comput. Struct. Biotechnol. J. 2020, 18, 2000–2011. [Google Scholar]
- Cerosaletti, K.; Barahmand-pour-Whitman, F.; Yang, J.; DeBerg, H.A.; Dufort, M.J.; Murray, S.A.; Israelsson, E.; Speake, C.; Gersuk, V.H.; Eddy, J.A.; et al. Single-cell RNA sequencing reveals expanded clones of islet antigen-reactive CD4 T cells in peripheral blood of subjects with type 1 diabetes. J. Immunol. 2017, 199, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, K.; Shoda, H.; Kochi, Y.; Yasui, T.; Kadono, Y.; Tanaka, S.; Fujio, K.; Yamamoto, K. Quantitative and qualitative characterization of expanded CD4+ T cell clones in rheumatoid arthritis patients. Sci. Rep. 2015, 5, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon, D.; Bhaskar, A.; Knowles, D.A.; Golan, D.; Raj, T.; Fu, A.Q.; Pritchard, J.K. Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet. 2017, 101, 686–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ENCODE Project Consortium The ENCODE (ENCyclopedia of DNA elements) project. Science 2004, 306, 636–640. [CrossRef] [PubMed] [Green Version]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.M.; de la Torre, V.; Richardson, D.; Royo, R.; Puiggròs, M.; Moncunill, V.; Fragkogianni, S.; Clarke, L.; BLUEPRINT Consortium; Flicek, P.; et al. The BLUEPRINT data analysis portal. Cell Syst. 2016, 3, 491–495.e5. [Google Scholar]
- Angiolilli, C.; Marut, W.; van der Kroef, M.; Chouri, E.; Reedquist, K.A.; Radstake, T.R.D.J. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 2018, 14, 657–673. [Google Scholar] [CrossRef]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Nordmark, G. Epigenetic alterations in primary Sjögren’s syndrome—An overview. Clin. Immunol. 2018, 196, 12–20. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Ota, M.; Sumitomo, S.; Ishigaki, K.; Suzuki, A.; Sakata, T.; Tsuchida, Y.; Inui, H.; Hirose, J.; Kochi, Y.; et al. Parsing multiomics landscape of activated synovial fibroblasts highlights drug targets linked to genetic risk of rheumatoid arthritis. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Pelikan, R.C.; Kelly, J.A.; Fu, Y.; Lareau, C.A.; Tessneer, K.L.; Wiley, G.B.; Wiley, M.M.; Glenn, S.B.; Harley, J.B.; Guthridge, J.M.; et al. Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks. Nat. Commun. 2018, 9, 2905. [Google Scholar] [CrossRef]
- Kong, X.; Sawalha, A.H. Takayasu arteritis risk locus in represses the anti-inflammatory gene through chromatin looping and recruiting MEF2-HDAC complex. Ann. Rheum. Dis. 2019, 78, 1388–1397. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ’t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.-J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odhams, C.A.; Cunninghame Graham, D.S.; Vyse, T.J. Profiling RNA-Seq at multiple resolutions markedly increases the number of causal eQTLs in autoimmune disease. PLoS Genet. 2017, 13, e1007071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorkin, D.U.; Qiu, Y.; Hu, M.; Fletez-Brant, K.; Liu, T.; Schmitt, A.D.; Noor, A.; Chiou, J.; Gaulton, K.J.; Sebat, J.; et al. Common DNA sequence variation influences 3-dimensional conformation of the human genome. Genome Biol. 2019, 20, 255. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; McGovern, A.; Orozco, G.; Duffus, K.; Yarwood, A.; Schoenfelder, S.; Cooper, N.J.; Barton, A.; Wallace, C.; Fraser, P.; et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun. 2015, 6, 10069. [Google Scholar] [CrossRef] [Green Version]
- McGovern, A.; Schoenfelder, S.; Martin, P.; Massey, J.; Duffus, K.; Plant, D.; Yarwood, A.; Pratt, A.G.; Anderson, A.E.; Isaacs, J.D.; et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol. 2016, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Ding, J.; Duffus, K.; Gaddi, V.P.; McGovern, A.; Ray-Jones, H.; Yarwood, A.; Worthington, J.; Barton, A.; Orozco, G. Chromatin interactions reveal novel gene targets for drug repositioning in rheumatic diseases. Ann. Rheum. Dis. 2019, 78, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Schoenfelder, S.; Furlan-Magaril, M.; Mifsud, B.; Tavares-Cadete, F.; Sugar, R.; Javierre, B.-M.; Nagano, T.; Katsman, Y.; Sakthidevi, M.; Wingett, S.W.; et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015, 25, 582–597. [Google Scholar] [CrossRef] [Green Version]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef] [Green Version]
- Burren, O.S.; Rubio García, A.; Javierre, B.-M.; Rainbow, D.B.; Cairns, J.; Cooper, N.J.; Lambourne, J.J.; Schofield, E.; Castro Dopico, X.; Ferreira, R.C.; et al. Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol. 2017, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Pradhananga, S.; Spalinskas, R.; Poujade, F.-A.; Eriksson, P.; Sahlén, P. Promoter anchored interaction landscape of THP-1 macrophages captures early immune response processes. Cell. Immunol. 2020, 355, 104148. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017, 49, 1602–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasolino, M.; Goldman, N.; Wang, W.; Cattau, B.; Zhou, Y.; Petrovic, J.; Link, V.M.; Cote, A.; Chandra, A.; Silverman, M.; et al. Genetic variation in type 1 diabetes reconfigures the 3D chromatin organization of T cells and alters gene expression. Immunity 2020, 52, 257–274.e11. [Google Scholar] [CrossRef]
- Pasula, S.; Tessneer, K.L.; Fu, Y.; Gopalakrishnan, J.; Pelikan, R.C.; Kelly, J.A.; Wiley, G.B.; Wiley, M.M.; Gaffney, P.M. Role of systemic lupus erythematosus risk variants with opposing functional effects as a driver of hypomorphic expression of TNIP1 and other genes within a three-dimensional chromatin network. Arthritis Rheumatol. 2020, 72, 780–790. [Google Scholar] [CrossRef]
- Shi, C.; Ray-Jones, H.; Ding, J.; Duffus, K.; Fu, Y.; Gaddi, V.P.; Gough, O.; Hankinson, J.; Martin, P.; McGovern, A.; et al. An active chromatin interactome in relevant cell lines elucidates biological mechanisms at genetic risk loci for dermatological traits. BioRxiv 2020. [Google Scholar] [CrossRef]
- Bourges, C.; Groff, A.F.; Burren, O.S.; Gerhardinger, C.; Mattioli, K.; Hutchinson, A.; Hu, T.; Anand, T.; Epping, M.W.; Wallace, C.; et al. Resolving mechanisms of immune-mediated disease in primary CD4 T cells. EMBO Mol. Med. 2020, 12, e12112. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yang, S.; Yu, D.; Gao, W.; Liu, X.; Zhang, K.; Fu, X.; Bao, W.; Zhang, K.; Yu, J.; et al. CRISPR/cas9 mediated knockout of an intergenic variant rs6927172 identified IL-20RA as a new risk gene for multiple autoimmune diseases. Genes Immun. 2018, 20, 103–111. [Google Scholar] [CrossRef]
- Shifrut, E.; Carnevale, J.; Tobin, V.; Roth, T.L.; Woo, J.M.; Bui, C.T.; Li, P.J.; Diolaiti, M.E.; Ashworth, A.; Marson, A. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 2018, 175, 1958–1971.e15. [Google Scholar] [CrossRef] [Green Version]
- Thynn, H.N.; Chen, X.-F.; Hu, W.-X.; Duan, Y.-Y.; Zhu, D.-L.; Chen, H.; Wang, N.-N.; Chen, H.-H.; Rong, Y.; Lu, B.-J.; et al. An allele-specific functional SNP associated with two systemic autoimmune diseases modulates IRF5 expression by long-range chromatin loop formation. J. Investig. Dermatol. 2020, 140, 348–360.e11. [Google Scholar] [CrossRef] [PubMed]
- Giménez, C.A.; Ielpi, M.; Mutto, A.; Grosembacher, L.; Argibay, P.; Pereyra-Bonnet, F. CRISPR-on system for the activation of the endogenous human INS gene. Gene Ther. 2016, 23, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.-S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a patient with SCID: Essential role of jak3 in lymphoid development. Science 1995, 270, 797–800. [Google Scholar] [CrossRef] [Green Version]
- Van Vollenhoven, R.F.; Fleischmann, R.; Cohen, S.; Lee, E.B.; García Meijide, J.A.; Wagner, S.; Forejtova, S.; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsmore, K.M.; Grammer, A.C.; Lipsky, P.E. Drug repurposing to improve treatment of rheumatic autoimmune inflammatory diseases. Nat. Rev. Rheumatol. 2020, 16, 32–52. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; ULTRA-DD Consortium; De Wolf, H.; Knezevic, B.; Burnham, K.L.; Osgood, J.; Sanniti, A.; LledóLara, A.; Kasela, S.; De Cesco, S.; et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 2019, 51, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Chen, L.; Knight, J.C. From genome-wide association studies to rational drug target prioritisation in inflammatory arthritis. Lancet Rheumatol. 2020, 2, e50–e62. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Serna, D.; Villanueva-Martin, G.; Acosta-Herrera, M.; Márquez, A.; Martín, J. Approaching Shared Pathophysiology in Immune-Mediated Diseases through Functional Genomics. Genes 2020, 11, 1482. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121482
González-Serna D, Villanueva-Martin G, Acosta-Herrera M, Márquez A, Martín J. Approaching Shared Pathophysiology in Immune-Mediated Diseases through Functional Genomics. Genes. 2020; 11(12):1482. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121482
Chicago/Turabian StyleGonzález-Serna, David, Gonzalo Villanueva-Martin, Marialbert Acosta-Herrera, Ana Márquez, and Javier Martín. 2020. "Approaching Shared Pathophysiology in Immune-Mediated Diseases through Functional Genomics" Genes 11, no. 12: 1482. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121482