Improved Genetic Map Identified Major QTLs for Drought Tolerance- and Iron Deficiency Tolerance-Related Traits in Groundnut

 ,
,  ,
,  ,
,  ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Phenotyping for Drought Tolerance- and Iron Deficiency (ID) Tolerance-Related Traits
2.2. DNA Extraction and Genotyping with 58K SNPs “Axiom_Arachis” Array
2.3. SNP Allele Calling, Filtering, and Quality Control
2.4. Construction of Genetic Map
2.5. Identification of Major Main-Effect and Epistatic QTLs
3. Results
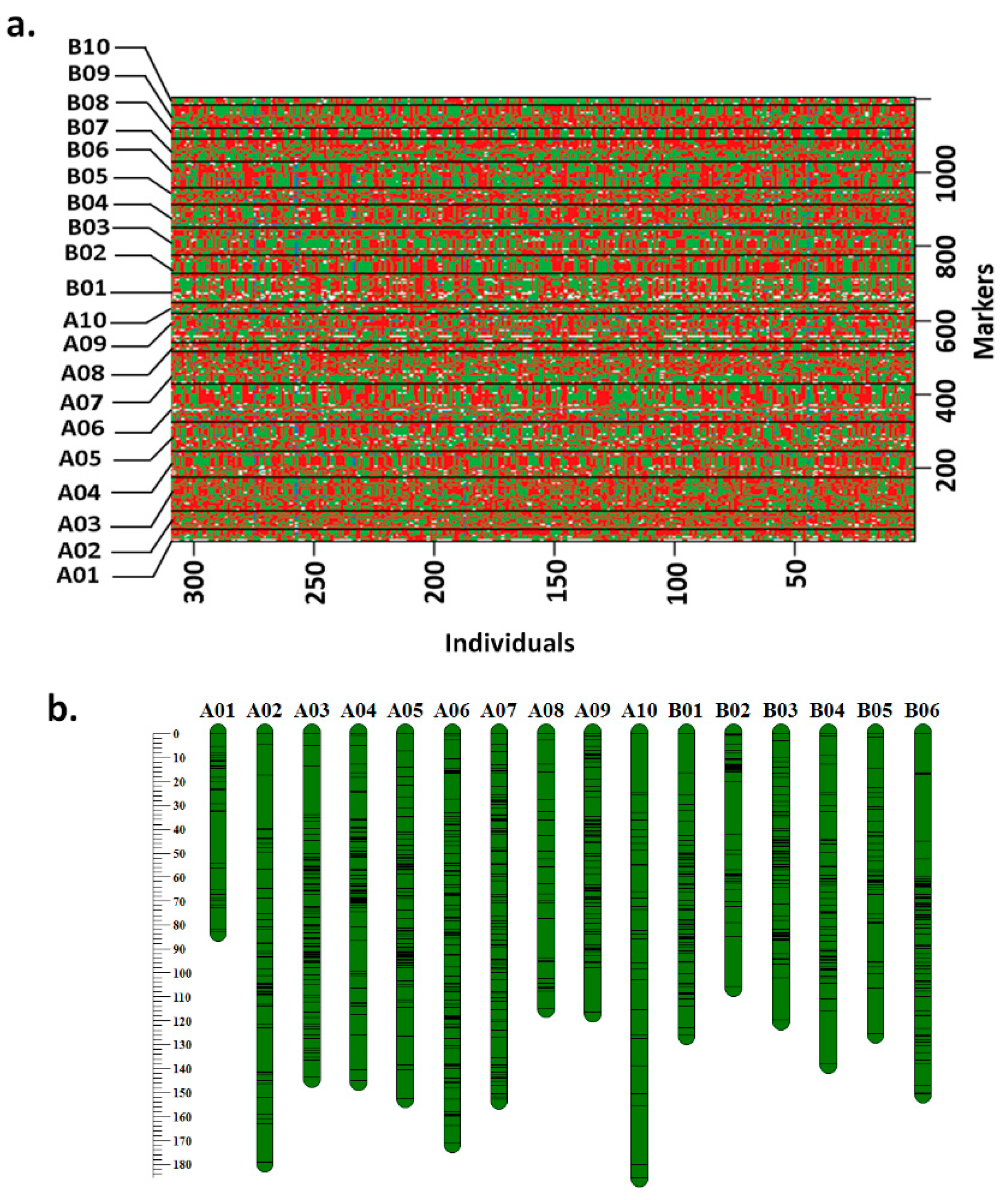
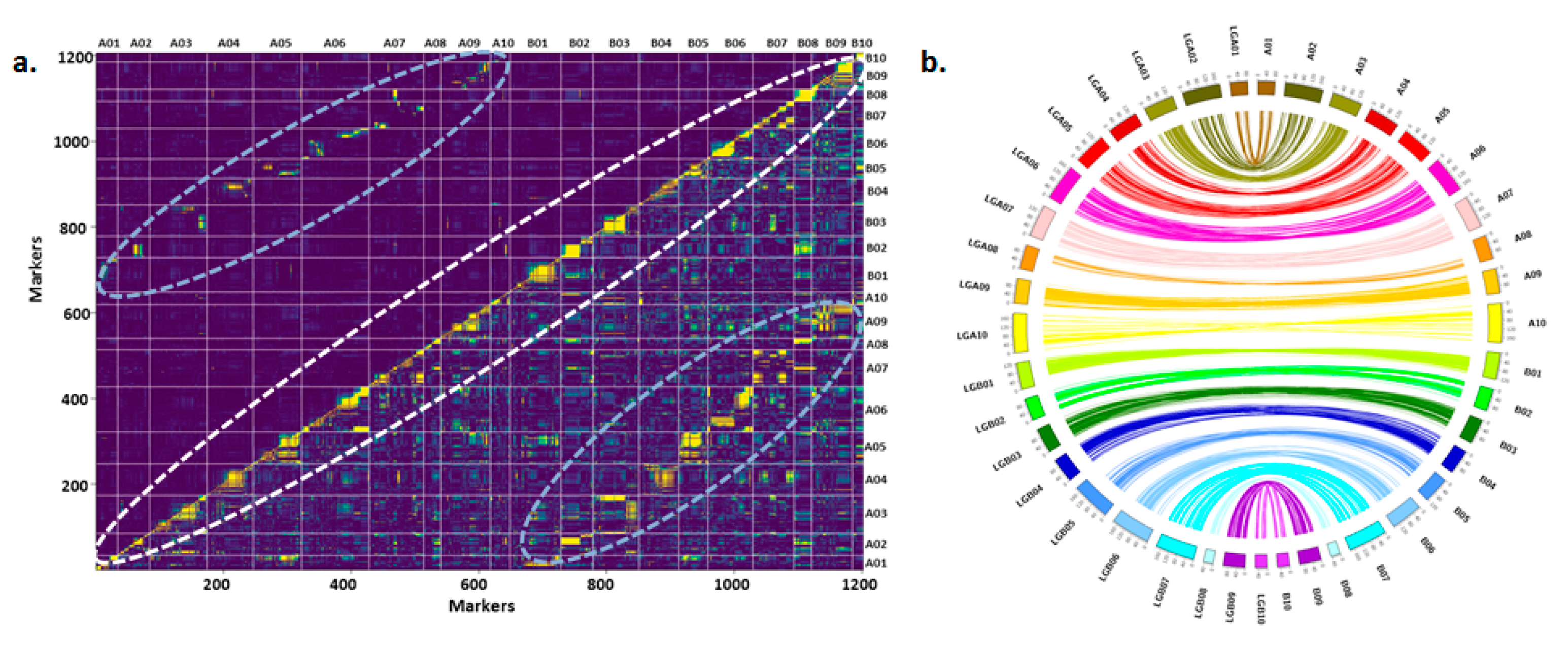
3.1. Refined Genetic Map on RIL Population TAG 24 × ICGV 86031
3.2. Genome-Wide Main-Effect QTLs for Drought Tolerance- and Iron Deficiency (ID) Tolerance-Related Traits
3.2.1. Main-Effect QTLs for Drought Tolerance-Related Traits
3.2.2. QTLs for ID Tolerance-Related Traits
3.3. Epistatic Interactions (E-QTLs) for Drought Tolerance- and ID Tolerance-Related Traits
3.3.1. E-QTLs for Drought Tolerance-Related Traits
3.3.2. E-QTLs for ID Tolerance-Related Traits
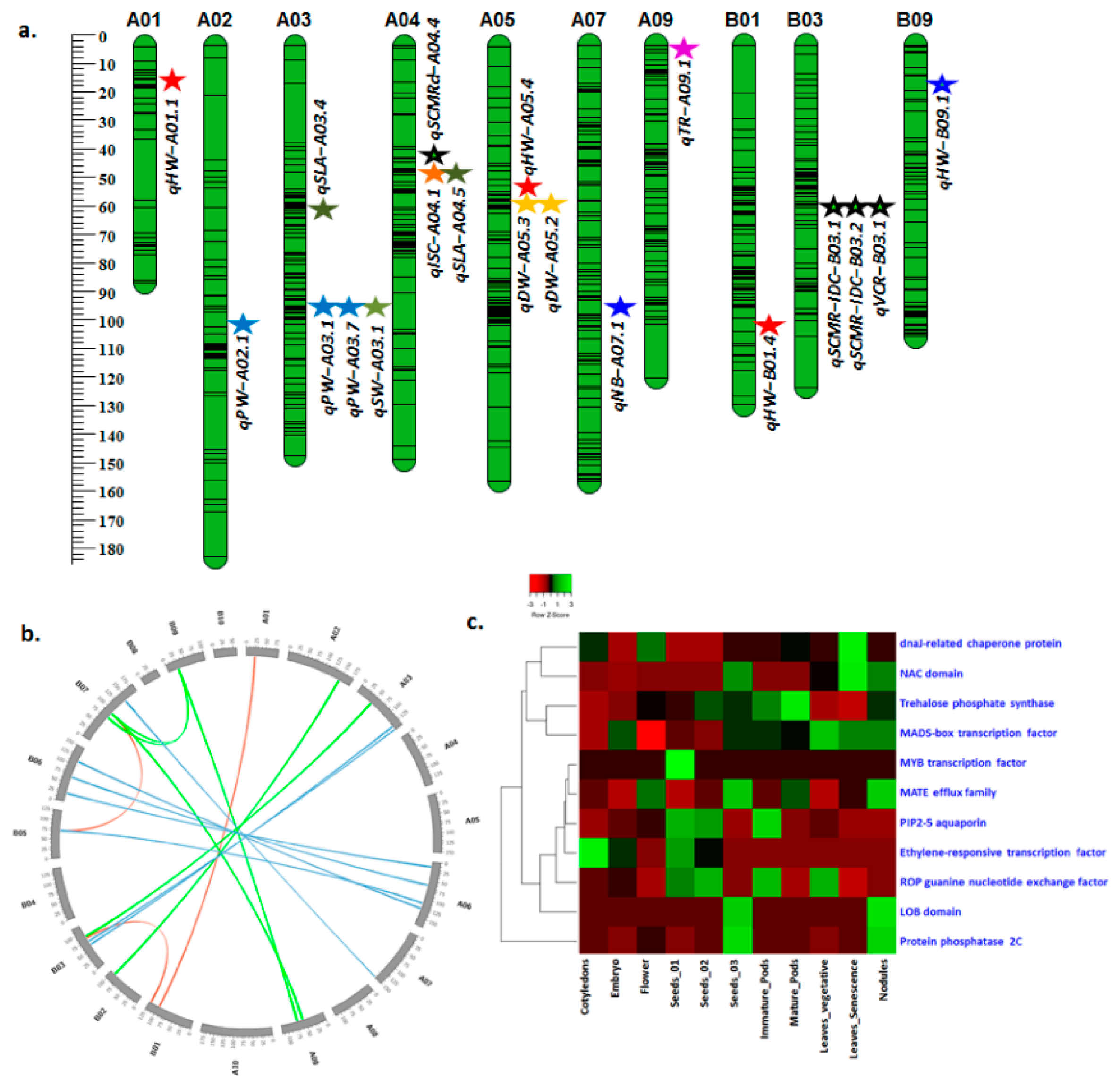
3.4. Candidate Genes Underlying Major QTL Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QTL | Quantitative trait locus |
| PVE | Phenotypic variation explained |
| GBS | Genotyping by sequencing |
| WGRS | Whole-genome re-sequencing |
| ddRAD-seq | Double digest restriction-site associated DNA |
| LOD | Logarithm of the odds |
| cM | CentiMorgan |
| Lob | Lateral organ boundaries |
| ABA | Abscisic acid |
| GH | Glycosyl hydrolase |
| LEA | Late embryogenesis abundant |
| bHLH | Basic helix-loop-helix |
| LRR | Leucine-rich repeat |
| NAC | NAM, ATAF, and CUC |
| ROP | Rho of plants |
| MAS | Marker-assisted selection |
| MYB | Myeloblastosis |
References
- Food and Agriculture Organization of the United Nations Database (FAOSTAT). 2020. Available online: http://faostat.fao.org/database (accessed on 13 April 2020).
- Lesk, C.; Rowhani, P.; Ramankutty, N. Influence of extreme weather disasters on global crop production. Nature 2016, 529, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangurde, S.S.; Kumar, R.; Pandey, A.K.; Burow, M.; Laza, H.E.; Nayak, S.N.; Guo, B.; Liao, B.; Bhat, R.S.; Madhuri, N.; et al. Climate-smart groundnuts for achieving high productivity and improved quality: Current status, challenges, and opportunities. In Genomic Designing of Climate-Smart Oilseed Crops; Kole, C., Ed.; Springer: Cham, Switzerland, 2019; pp. 133–172. [Google Scholar]
- Wright, G.C.; Nageswara Rao, R.C. Peanut Water Relations. In The Peanut Crop; Smartt, J., Ed.; Chapman and Hall: London, UK, 1994; pp. 281–325. [Google Scholar]
- Barnabas, B.; Jäger, K.; Fehér, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, I. Plant nutrition research: Priorities to meet human needs for food in sustainable way. Plant Soil 2002, 247, 3–24. [Google Scholar] [CrossRef]
- Singh, A.L. Yield losses in groundnut due to micronutrient deficiencies in calcareous soils of India. Plant Nutr. 2001, 92, 838–839. [Google Scholar] [CrossRef]
- Kihara, J.; Bolo, P.; Kinyua, M.; Rurinda, J.; Piikki, K. Micronutrient deficiencies in African soils and the human nutritional nexus: Opportunities with staple crops. Environ. Geochem. Health 2020, 42, 3015–3033. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Pandey, A.K.; Kumar, R.; Nwosu, V.; Guo, B.; Wright, G.; Bhat, R.S.; Chen, X.; Bera, S.K.; Yuan, M.; et al. Translational genomics for achieving higher genetic gains in groundnut. Theor. Appl. Genet. 2020, 133, 1679–1702. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Thudi, M.; Pandey, M.K.; Tardieu, F.; Ojiewo, C.; Vadez, V.; Whitbread, A.M.; Siddique, K.H.M.; Nguyen, H.T.; Carberry, P.S.; et al. Accelerating genetic gains in legumes for prosperous smallholder agriculture: Integrating genomics, phenotyping, systems modelling and agronomy. J. Exp. Bot. 2018, 69, 3293–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Chaudhari, S.; Jarquin, D.; Janila, P.; Crossa, J.; Patil, S.C.; Sundravadana, S.; Khare, D.; Bhat, R.S.; Radhakrishnan, T.; et al. Genome-based trait prediction in multi- environment breeding trials in groundnut. Theor. Appl. Genet. 2020, 133, 3101–3117. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Bertioli, D.J.; Moretzohn, M.C.; Vadez, V.; Krishnamurty, L.; Aruna, R.; Nigam, S.N.; Ravi, K.; He, G.; Knapp, S.J.; et al. The first SSR based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 2009, 118, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Ribaut, J.M.; Buckler, E.S.; Tuberosa, R.; Rafalski, J.A.; Langridge, P. Can genomics boost productivity of orphan crops? Nat. Biotechnol. 2012, 30, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Gautami, B.; Pandey, M.K.; Vadez, V.; Nigam, S.N.; Ratnakumar, P.; Krishnamurthy, L.; Radhakrishnan, T.; Gowda, M.V.; Narasu, M.L.; Hoisington, D.A.; et al. Quantitative trait locus analysis, and construction of consensus genetic map for drought tolerance related traits based on three recombinant inbred line populations in cultivated groundnut (Arachis hypogaea L.). Mol. Breed. 2012, 30, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautami, B.; Foncéka, D.; Pandey, M.K.; Moretzsohn, M.C.; Sujay, V.; Qin, H.; Hong, Y.; Faye, I.; Chen, X.; BhanuPrakash, A.; et al. An international reference consensus genetic map with 897 marker loci based on 11 mapping populations for tetraploid groundnut (Arachis hypogaea L.). PLoS ONE 2012, 7, e41213. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Penmetsa, R.V.; Yuan, M.; Gong, L.; Zhao, Y.; Guo, B.; Farmer, A.D.; Rosen, B.D.; Gao, J.; Isobe, S.; et al. Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (Arachis hypogaea L.). BMC Plant Biol. 2012, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ravi, K.; Vadez, V.; Isobe, S.; Mir, R.; Guo, Y.; Nigam, S.N.; Gowda, M.V.C.; Radhakrishnan, T.; Bertioli, D.J.; Knapp, S.J.; et al. Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor. Appl. Genet. 2011, 122, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Faye, I.; Pandey, M.K.; Hamidou, F.; Rathore, A.; Ndoye, O.; Vadez, V.; Varshney, R.K. Identification of quantitative trait loci for yield and yield related traits in groundnut (Arachis hypogaea L.) under different water regimes in Niger and Senegal. Euphytica 2015, 3, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jiao, Y.; Cheng, L.; Huang, L.; Liao, B.; Tang, M.; Ren, X.; Zhou, X.; Chen, Y.; Jiang, H. Quantitative trait locus analysis for pod-and kernel-related traits in the cultivated peanut (Arachis hypogaea L.). BMC Genet. 2016, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Pattanashetti, S.K.; Pandey, M.K.; Naidu, G.K.; Vishwakarma, M.K.; Singh, O.K.; Shasidhar, Y.; Boodi, I.H.; Biradar, B.D.; Das, R.R.; Rathore, A.; et al. Identification of quantitative trait loci associated with iron deficiency chlorosis resistance in groundnut (Arachis hypogaea L.). Plant Breed. 2020, 139, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Agarwal, G.; Kale, S.M.; Clevenger, J.; Nayak, S.N.; Sriswathi, M.; Chitikineni, A.; Chavarro, C.; Chen, X.; Upadhyaya, H.D.; et al. Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58K SNPs for accelerating genetics and breeding in groundnut. Sci. Rep. 2017, 7, 40577. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Gao, D.; Dudchenko, O.; Seijo, G.; Leal-Bertioli, S.; Ren, L.; Farmer, A.; Pandey, M.K.; et al. The genome sequence of peanut (Arachis hypogaea L.), a segmental allotetraploid. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, Q.; Liu, H.; Zhang, J.; Hong, Y.; Lan, H.; Li, H.; Wang, J.; Liu, H.; Li, S.; et al. Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant 2019, 12, 920–934. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The Arachis hypogaea genome elucidates legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–887. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Khan, A.W.; Singh, V.K.; Vishwakarma, M.K.; Shasidhar, Y.; Kumar, V.; Garg, V.; Bhat, R.S.; Chitikineni, A.; Janila, P.; et al. QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.). Plant Biotechnol. J. 2017, 15, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, L.; Vadez, V.; Jyotsna Devi, M.; Serraj, R.; Nigam, S.N.; Sheshshayee, M.S.; Chandra, S.; Aruna, R. Variation in transpiration efficiency and its related traits in a groundnut (Arachis hypogaea L.) mapping population. Field Crops Res. 2007, 103, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Naidu, G.K.; Pattanashetti, S.K.; Boodi, I.H.; Omprakash Kumar, S.; Prakyath Kumar, K.V.; Biradar, B.D.; Wali, M.C. Genetic analysis of recombinant inbred lines for iron deficiency chlorosis and productivity traits in groundnut. Indian J. Genet. Plant Breed. 2017, 77, 414–421. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap 4. In Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma, B.V., Ed.; Plant Research International: Wageningen, The Netherlands, 2006. [Google Scholar]
- Voorrips, R.E. Mapchart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Bajaj, P.; Pazhamala, L.; Nayak, S.; Pandey, M.K.; Chitikineni, A.; Huai, D.; Khan, A.W.; Desai, A.; Jiang, H.; et al. The Arachis hypogaea gene expression atlas (AhGEA) for accelerating translational research in cultivated groundnut. Plant Biotechnol. J. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohra, A.; Jha, U.C.; Godwin, I.D.; Varshney, R.K. Genomic interventions for sustainable agriculture. Plant Biotechnol. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Janila, P.; Pandey, M.K.; Shasidhar, Y.; Variath, M.T.; Sriswathi, M.; Khera, P.; Manohar, S.S.; Nagesh, P.; Vishwakarma, M.K.; Mishra, G.P.; et al. Molecular breeding for introgression of fatty acid desaturase mutant alleles (ahFAD2A and ahFAD2B) enhances oil quality in high and low oil containing peanut genotypes. Plant Sci. 2016, 242, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Bera, S.K.; Kamdar, J.H.; Kasundra, S.V.; Dash, P.; Maurya, A.K.; Jasani, M.D.; Chandrashekar, A.B.; Manivannan, N.; Vasanthi, R.P.; Dobariya, K.L.; et al. Improving oil quality by altering levels of fatty acids through marker-assisted selection of ahfad2 alleles in peanut (Arachis hypogaea L.). Euphytica 2018, 214, 162. [Google Scholar] [CrossRef]
- Varshney, R.K.; Pandey, M.K.; Janila, P.; Nigam, S.N.; Sudini, H.; Gowda, M.V.; Sriswathi, M.; Radhakrishnan, T.; Manohar, S.S.; Nagesh, P. Marker-assisted introgression of a QTL region to improve rust resistance in three elite and popular varieties of peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2014, 127, 1771–1781. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Shi, Y. Genetic differences in resistance to iron deficiency chlorosis in peanut. J. Plant Nutr. 2007, 30, 37–52. [Google Scholar] [CrossRef]
- Akhtar, S.; Shahzad, A.; Arshad, M.; Hassan, F. Morpho-physiological evaluation of groundnut (Arachis hypogaea L.) genotypes for iron deficiency tolerance. Pak. J. Bot. 2013, 45, 893–899. [Google Scholar]
- King, K.E. Mineral Evaluation and Quantitative Trait Loci Mapping in a Soybean [Glycine max (L.)Merr.] Population Developed for Iron Deficiency Chlorosis. Paper 10298. Bachelor’s Thesis, Iowa State University, Iowa, IA, USA, 2011. [Google Scholar]
- Pandey, M.K.; Monyo, E.; Ozias-Akins, P.; Liang, X.; Guimarães, P.; Nigam, S.N.; Upadhyaya, H.D.; Janila, P.; Zhang, X.; Guo, B.; et al. Advances in Arachis genomics for peanut improvement. Biotechnol. Adv. 2012, 30, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Fang, Y.; Hu, P.; Tan, Y.; Wang, Y.; Hou, L.; Deng, X.; Wu, H.; Zhu, L.; Zhu, L.; et al. Construction of a high-density genetic map based on SLAF markers and QTL analysis of leaf size in rice. Front. Plant Sci. 2020, 11, 1143. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, X.; Miao, H.; Chu, Y.; Cui, F.; Yang, W.; Wang, C.; Shen, Y.; Xu, T.; Zhao, L.; et al. QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 2019, 19, 537. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Chen, H.; Deng, Y.; Chen, Y.; Zhang, C.; Cai, T.; Ali, N.; Mamadou, G.; Xie, D.; Guo, B.; et al. High-density SNP map facilitates fine mapping of QTLs and candidate genes discovery for Aspergillus flavus resistance in peanut (Arachis hypogaea). Theor. Appl. Genet. 2020, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.H.; Zhang, S.Z.; Miao, H.R.; Cui, F.G.; Shen, Y.; Yang, W.Q.; Xu, T.T.; Chen, N.; Chi, X.Y.; Zhang, Z.M.; et al. High-density genetic map construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Negro, S.S.; Millet, E.J.; Madur, D.; Bauland, C.; Combes, V.; Welcker, C.; Tardieu, F.; Charcosset, A.; Nicolas, S.D. Genotyping-by-sequencing and SNP-arrays are complementary for detecting quantitative trait loci by tagging different haplotypes in association studies. BMC Plant Biol. 2019, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- Ribaut, J.M.; Jiang, C.; Gonzalez-de-Leon, D.; Edmeades, G.O.; Hoisington, D.A. Identification of quantitative trait loci under drought conditions in tropical maize. 2. Yield components and marker-assisted selection strategies. Theor. Appl. Genet. 1997, 94, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.S.; Liu, F.; Cao, J.; Yue, B.; Xiong, L. Detecting quantitative trait loci for water use efficiency in rice using a recombinant inbred line population. Chin. Sci. Bull. 2011, 56, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Suji, K.K.; Biji, K.R.; Poornima, R.; Prince, K.S.J.; Amudha, K.; Kavitha, S.; Mankar, S.; Babu, R.C. Mapping QTLs for plant phenology and production traits using indica rice (Oryza sativa L.) lines adapted to rainfed environment. Mol. Biotechnol. 2012, 52, 151–160. [Google Scholar] [CrossRef]
- Sharma, V.; Verma, R.K.; Dey, P.C.; Chetia, S.K.; Baruah, A.R.; Modi, M.K. QTLs associated with yield attributing traits under drought stress in upland rice cultivar of Assam. Oryza 2017, 54, 253–257. [Google Scholar] [CrossRef]
- Martin, I.; Tenorio, J.L.; Ayerbe, L. Yield, growth and water use of conventional and semi-leafless peas in semiarid environments. Crop Sci. 1994, 34, 1576–1583. [Google Scholar] [CrossRef]
- Yu, S.B.; Li, J.X.; Xu, C.G.; Tan, Y.F.; Gao, Y.J.; Li, X.H.; Zhang, Q.; Maroof, M.A.S. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 1997, 94, 9226–9231. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Sandhu, N.; Majumder, R.R.; Dixit, S.; Kumar, S.; Singh, S.P.; Mandal, N.P.; Das, S.P.; Yadaw, R.B.; Singh, V.K.; et al. Epistatic interactions of major effect drought QTLs with genetic background loci determine grain yield of rice under drought stress. Sci. Rep. 2019, 9, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, N.; Dixit, S.; Swamy, B.M.; Vikram, P.; Venkateshwarlu, C.; Catolos, M.; Kumar, A. Positive interactions of major-effect QTLs with genetic background that enhances rice yield under drought. Sci. Rep. 2018, 8, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodia, S.M.; Joshi, B.; Gangurde, S.S.; Thirumalaisamy, P.P.; Mishra, G.P.; Narandrakumar, D.; Soni, P.; Rathnakumar, A.L.; Dobaria, J.R.; Sangh, C.; et al. Genotyping-by-sequencing based genetic mapping reveals large number of epistatic interactions for stem rot resistance in groundnut. Theor. Appl. Genet. 2019, 132, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Xie, G.; Yang, B.; Xu, Z.; Li, F.; Guo, K.; Zhang, M.; Wang, L.; Zou, W.; Wang, Y.; Peng, L. Global identification of multiple OsGH9 family members and their involvement in cellulose crystallinity modification in rice. PLoS ONE 2013, 8, e50171. [Google Scholar] [CrossRef]
- Szyjanowicz, P.M.; McKinnon, I.; Taylor, N.G.; Gardiner, J.; Jarvis, M.C.; Turner, S.R. The irregular xylem 2 mutant is an allele of korrigan that affects the secondary cell wall of Arabidopsis thaliana. Plant J. 2004, 37, 730–740. [Google Scholar] [CrossRef]
- Lopez-Casado, G.; Urbanowicz, B.R.; Damasceno, C.M.; Rose, J.K. Plant glycosyl hydrolases and biofuels: A natural marriage. Curr. Opin. Plant Biol. 2008, 11, 329–337. [Google Scholar] [CrossRef]
- Carrari, F.; Loureiro, M.E.; Ratcliffe, R.G.; Sweetlove, L.J.; Fernie, A.R. Enhanced photosynthetic performance and growth as a consequence of decreasing mitochondrial malate dehydrogenase activity in transgenic tomato plants. Plant Physiol. 2005, 137, 611–622. [Google Scholar]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P. The basic helix-loop-helix transcription factor family in plants A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, J.; He, X.; Wang, Y.; Ma, X.; Yin, D. Genome-wide association study of major agronomic traits related to domestication in peanut. Front. Plant Sci. 2017, 8, 1611. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Ono, E.; Mizutani, M. Evolution and diversity of the 2-oxoglutarate-dependent dioxygenase superfamily in plants. Plant J. 2014, 78, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.B.; Giehl, R.F.; Doll, S.; Mock, H.P.; Strehmel, N.; Scheel, D.; Kong, X.; Hider, R.C.; von Wirén, N. Feruloyl-CoA 6ʹ-Hydroxylase1-dependent coumarins mediate iron acquisition from alkaline substrates in Arabidopsis. Plant Physiol. 2014, 164, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.J.; Lin, W.D.; Schmidt, W. Transcriptional profiling of the Arabidopsis iron deficiency response reveals conserved transition metal homeostasis networks. Plant Physiol. 2010, 152, 2130–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, R.J.; Waters, B.M. Use of natural variation reveals core genes in the transcriptome of iron-deficient Arabidopsis thaliana roots. J. Exp. Bot. 2012, 63, 1039–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.M.; Hindt, M.N.; Schmidt, H.; Clemens, S.; Guerinot, M.L. MYB10 and MYB72 are required for growth under iron-limiting conditions. PLoS Genet. 2013, 9, e1003953. [Google Scholar] [CrossRef] [Green Version]
- Zamioudis, C.; Korteland, J.; Van Pelt, J.A.; van Hamersveld, M.; Dombrowski, N.; Bai, Y.; Hanson, J.; Van Verk, M.C.; Ling, H.Q.; Schulze-Lefert, P.; et al. Rhizobacterial volatiles and photosynthesis-related signals coordinate MYB72 expression in Arabidopsis roots during onset of induced systemic resistance and iron-deficiency responses. Plant J. 2015, 84, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Stringlis, I.A.; Yua, K.; Feussnerb, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.A.; Feussner, I.; Pieterse, C.M. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.W.; Gregersen, P.L. Members of the barley NAC transcription factor gene family show differential co-regulation with senescence-associated genes during senescence of flag leaves. J. Exp. Bot. 2014, 65, 4009–4022. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Nam, H.G.; Lim, P.O. Regulatory network of NAC transcription factors in leaf senescence. Curr. Opin. Plant Biol. 2016, 33, 48–56. [Google Scholar] [CrossRef]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC proteins: Regulation and role in stress tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.S.; Kim, Y.S.; Redillas, M.C.; Jang, G.; Jung, H.; Bang, S.W.; Choi, Y.D.; Ha, S.H.; Reuzeau, C.; Kim, J.K. OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 2013, 11, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC Gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Derkx, A.P.; Liu, D.C.; Buchner, P.; Hawkesford, M.J. Overexpression of a NAC transcription factor delays leaf senescence and increases grain nitrogen concentration in wheat. Plant Biol. 2015, 17, 904–913. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Traits | 2004 | 2005 | 2008 | 2009 | 2010 | 2013 | 2014 | 2015 |
|---|---|---|---|---|---|---|---|---|
| Days to 50% flowering | - | - | - | - | - | - | PT | PT |
| Dry matter | - | PT (2) | - | - | - | - | - | - |
| Carbon discrimination ratio (delta13C) | PT | - | - | - | - | - | - | - |
| Haulm weight | PT (4) | - | PT-WW, PT-WS | SD-WS, SD-WW, BM-WW, BM-WS, PT-WW | SD-WW, SD-WS | PT-WW, PT-WS | PT-WW, PT-WS, | PT-WS, PT-WW |
| Harvest index | - | - | - | BM-WW, BM-WS, SD-WW, SD-WS, | SD-WW, SD-WS | - | PT-WW, PT-WS | - |
| 100 seed weight | - | - | PT-WW, PT-WS | PT-WW | - | PT-WW, PT-WS | PT-WW, PT-WS | PT-WW, PT-WS |
| Canopy conductance | PT-WW | PT-WW | - | - | - | - | - | - |
| Leaf area | PT-WW, PT-WS, PT-H | PT-WW, PT-WS, PT-H | PT-WW, PT-WS | - | SD-WW, SD-WS | - | - | - |
| Leaf number | - | - | - | - | SD-WW | - | - | - |
| Days to maturity | - | - | - | BM-WW, BM-WS | - | - | - | - |
| Number of branches | - | - | - | BM-WW | - | - | - | - |
| Pod weight | - | - | PT-WW, PT-WS | BM-WW, BM-WS, SD-WW, SD-WS | SD-WW, SD-WS | PT-WW, PT-WS | PT-WW, PT-WS | - |
| Pod yield | - | - | - | - | - | - | PT-WW | PT-WW |
| SCMR-drought | - | PT(8)-WW | - | BM-WW, BM-WS, SD-WW, SD-WS | SD-WW, SD-WS | PT-WW, PT-WS | PT-WW, PT-WS | - |
| Shelling percentage | - | - | - | - | - | PT-WW, PT-WS | PT-WW, PT-WS | PT-WW, PT-WS |
| Shoot dry weight | - | - | PT(4)-WW, PT(4)-WS | BM-WW, BM-WS | - | - | - | - |
| Total dry matter | - | PT-WW | - | - | - | - | PT-WS | PT-WS |
| Transpiration efficiency | PT-WW, PT-WS | PT-WW, PT-WS | PT(4)-WW, PT(4)-WS | - | - | - | - | - |
| Transpiration rate | PT-WS | PT-WS | PT(4)-WW, PT(4)-WS | - | - | - | - | - |
| Water use efficiency | PT | - | - | - | - | - | - | - |
| Iron Deficiency Tolerance | ||||||||
| SCMR-ID | - | - | - | - | - | VJ (3) | VJ (3) | - |
| Visual chlorosis rating (VCR) | - | - | - | - | - | VJ (3) | VJ (3) | - |
| Chr | Total Loci | Total Mapped SNP Loci | SSR Loci | Map Length (cM) | Average Map Distance (cM/ Locus) |
|---|---|---|---|---|---|
| A01 | 33 | 21 | 12 | 82.7 | 2.5 |
| A02 | 51 | 41 | 10 | 179.1 | 3.5 |
| A03 | 89 | 73 | 16 | 143.7 | 1.6 |
| A04 | 73 | 54 | 19 | 145.0 | 2.0 |
| A05 | 76 | 60 | 16 | 152.3 | 2.0 |
| A06 | 105 | 86 | 19 | 171.2 | 1.6 |
| A07 | 86 | 56 | 30 | 152.8 | 1.8 |
| A08 | 27 | 14 | 13 | 114.7 | 4.2 |
| A09 | 78 | 68 | 10 | 116.4 | 1.5 |
| A10 | 30 | 26 | 4 | 185.4 | 6.2 |
| B01 | 80 | 58 | 22 | 126.0 | 1.6 |
| B02 | 50 | 50 | 0 | 105.8 | 2.1 |
| B03 | 73 | 73 | 0 | 119.7 | 1.6 |
| B04 | 62 | 57 | 5 | 138.0 | 2.2 |
| B05 | 45 | 45 | 0 | 125.4 | 2.8 |
| B06 | 71 | 71 | 0 | 150.5 | 2.1 |
| B07 | 64 | 64 | 0 | 183.4 | 2.9 |
| B08 | 28 | 28 | 0 | 46.1 | 1.6 |
| B09 | 64 | 63 | 1 | 102.1 | 1.6 |
| B10 | 20 | 20 | 0 | 57.9 | 2.9 |
| Total | 1205 | 1028 | 177 | 2598.3 | 2.2 |
| Main Effect QTLs | Epistatic QTLs (E-QTLs) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Traits | WW | WS | Total QTLs | LOD Range | PVE Range (%) | WW | WS | Total E-QTL | LOD Range | PVE Range (%) |
| A. Drought Tolerance-Related Traits | ||||||||||
| Days to 50% flowering | 3 | 0 | 3 | 3.1–3.9 | 5.3–6.8 | 1 | 0 | 1 | 5 | 11.9 |
| Haulm weight | 16 | 6 | 22 | 3.1–13.7 | 4.3–20.1 | 158 | 8 | 166 | 5.0–19.8 | 16.4–65.1 |
| Total dry matter | 6 | 0 | 6 | 3.3–5.3 | 4.8–13.9 | 0 | 21 | 21 | 5.0–11.1 | 23.9–66.2 |
| Harvest index | 1 | 0 | 1 | 3.5 | 5.3 | - | - | - | - | - |
| Canopy conductance | 6 | 0 | 6 | 3.1–13.5 | 3.1–17.2 | - | - | - | - | - |
| Leaf number | - | - | - | - | - | 50 | 0 | 50 | 5.1–20.1 | 18.7–34.9 |
| Number of branches | 2 | 0 | 2 | 4.8–15.5 | 6.4–23.3 | - | - | - | - | - |
| Pod weight | 9 | 2 | 9 | 3.1–6.5 | 5.1–14.2 | 1 | 4 | 5 | 5.1–5.3 | 10.0–33.8 |
| Transpiration efficiency | 11 | 2 | 13 | 3.1–4.8 | 4.9–8.6 | 1 | 0 | 1 | 5 | 11.5 |
| Shelling % | 2 | 0 | 2 | 3.1 | 4.8–5.8 | 2 | 69 | 71 | 5.0–15.0 | 26.9–53.2 |
| Shoot dry weight | 9 | 2 | 11 | 3.1–6.3 | 4.2–9.3 | 210 | 3 | 213 | 5.0–20.7 | 12.1–46.3 |
| Leaf area | 16 | 7 | 23 | 3.1–10.8 | 5.0–16.2 | 14 | 0 | 14 | 5.0–8.0 | 29.0–36.3 |
| Seed weight | 6 | 1 | 7 | 3.2–6.4 | 3.9–15.0 | - | - | - | - | - |
| Transpiration rate | 11 | 2 | 13 | 3.2–9.7 | 4.3–17.3 | 3 | 0 | 3 | 5.1–5.9 | 25.8–33.4 |
| SCMR-drought | 2 | 10 | 11 | 3.2–5.3 | 4.8–10.8 | 1 | 0 | 1 | 5.13 | 15.8 |
| Water use efficiency | - | - | - | 5 | 0 | 5 | 5.2–6.1 | 20.6–32.5 | ||
| B. Iron Deficiency Tolerance-Related Traits | ||||||||||
| SCMR-ID | 2 | 0 | 3 | 3.2–4.8 | 4.4–22.4 | 47 | 0 | 47 | 5.0–16.6 | 15.8–57.7 |
| VCR | 2 | 0 | 1 | 4.4 | 33.9 | 10 | 0 | 10 | 5.0–6.8 | 14.6–70.9 |
| Traits | WW/WS | Loc | Year | QTL Name | Chr | Pos (cM) | Left Marker | Right Marker | LOD | PVE (%) | Add |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. Drought Tolerance-Related Traits | |||||||||||
| Dry weight increase | WW | PT | 2005 | qDW-A05.3 | A05 | 55 | PM375 | A05_25039519 | 3.4 | 10.0 | 0.3 |
| Total dry matter | WW | PT | 2005 | qDW-A05.2 | A05 | 55 | PM375 | A05_25039519 | 4.9 | 13.9 | 0.5 |
| Haulm weight | WW | BM | 2009 | qHW-A01.1 | A01 | 23 | Seq13A10 | A01_96982501 | 13.7 | 20.1 | 1.5 |
| WW | BM | 2009 | qHW-A05.4 | A05 | 50 | GM2246 | A05_25200285 | 11.6 | 16.3 | 1.3 | |
| WW | BM | 2009 | qHW-B01.4 | B01 | 101 | B01_134144284 | B01_134275884 | 7.0 | 10.8 | −1.1 | |
| Delta biomass | WW | PT | 2004 | qHW-B09.1 | B09 | 22 | B09_145215085 | B09_16205676 | 4.4 | 12.0 | −0.2 |
| Canopy conductance | WW | PT | 2004 | qISC-A04.1 | A04 | 49 | A04_2540668 | Seq19H03 | 13.5 | 17.2 | −0.1 |
| Number of branches | WW | BM | 2009 | qNB-A07.1 | A07 | 99 | IPAHM689 | TC1A02 | 15.5 | 23.3 | 0.4 |
| Pod weight | WS | PT | 2008 | qPW-A03.1 | A03 | 92 | A03_101625507 | A03_25161497 | 3.7 | 10.0 | −31.1 |
| WW | SD | 2009 | qPW-A02.1 | A02 | 101 | Seq16C06 | A02_67418614 | 3.7 | 10.1 | −6.4 | |
| WW | PT | 2008 | qPW-A03.2 | A03 | 92 | A03_101625507 | A03_25161497 | 3.6 | 14.2 | −75.7 | |
| SCMR-drought | WW | PT | 2004 | qSCMRd-A04.4 | A04 | 46 | GM694 | A04_2540668 | 5.3 | 10.8 | −0.7 |
| Specific leaf area | WW | PT | 2004 | qSLA-A03.4 | A03 | 58 | GM660 | GM679 | 8.7 | 12.0 | −1.8 |
| WW | PT | 2004 | qSLA-A04.5 | A04 | 49 | A04_2540668 | Seq19H03 | 10.8 | 16.2 | 17.5 | |
| 100 Seed weight | WW | PT | 2008 | qSW-A03.1 | A03 | 92 | A03_101625507 | A03_25161497 | 3.9 | 15.0 | −56.3 |
| Transpiration rate | WS | PT | 2008 | qTR-A09.1 | A09 | 3 | A09_117790031 | A09_118421381 | 9.7 | 17.3 | −33.6 |
| B. Iron Deficiency Tolerance-Related Traits | |||||||||||
| VCR at 30DAS | WW | VJ | 2014 | qVCR-B03.1 | B03 | 62 | B03_13454528 | B03_10796590 | 4.4 | 33.9 | −0.3 |
| SCMR-ID at 60DAS | WW | VJ | 2014 | qSCMR-ID-B03.1 | B03 | 62 | B03_13454528 | B03_10796590 | 4.8 | 22.4 | 2.9 |
| SCMR-ID at 90DAS | WW | VJ | 2014 | qSCMR-ID-B03.2 | B03 | 62 | B03_13454528 | B03_10796590 | 4.8 | 11.0 | 2.7 |
| Trait | WW/WS | Loc | Year | QTL(s)_Name | Chr 1 | Pos 1 (cM) | Left Flanking Marker_1 | Right Flanking Marker_1 | Chr 2 | Pos 2 (cM) | Left Flanking Marker_2 | Right Flanking Marker_2 | LOD | PVE(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. Drought Tolerance-Related Traits | ||||||||||||||
| Haulm weight | WW | PT | 2008 | qqHAULM.5 | B07 | 75 | B07_1945835 | B07_1907545 | B09 | 25 | Seq19B01 | B09_16205676 | 5.49 | 36.7 |
| WW | PT | 2008 | qqHAULM.6 | B07 | 90 | B07_995898 | B07_578122 | B09 | 25 | Seq19B01 | B09_16205676 | 5.02 | 35.2 | |
| WW | PT | 2008 | qqHAULM.7 | A09 | 55 | A09_29837655 | A09_14222907 | B07 | 90 | B07_995898 | B07_578122 | 6.07 | 34.8 | |
| WW | PT | 2008 | qqHAULM.9 | A09 | 70 | A09_113636594 | A09_8787029 | B09 | 25 | Seq19B01 | B09_16205676 | 5.79 | 26.2 | |
| WW | PT | 2008 | qqHAULM.10 | A02 | 145 | A02_88924681 | A02_89185231 | B02 | 90 | B02_99720342 | B02_101402317 | 5.11 | 25.6 | |
| WW | PT | 2008 | qqHAULM.11 | A03 | 45 | A03_131260373 | IPAHM177 | B03 | 85 | B03_132031566 | B03_22451302 | 5.2 | 21.9 | |
| SCMR-drought | WW | PT | 2004 | qqSCMR.48 | A06 | 110 | A06_10555841 | A06_14851576 | B05 | 70 | B05_122054913 | B05_148647711 | 5.13 | 15.8 |
| B. Iron Deficiency Tolerance-Related Traits | ||||||||||||||
| SCMR-ID at 60DAS | WW | VJ | 2013 | qqSCMR.1 | A06 | 10 | A06_105316360 | A06_110918357 | B06 | 20 | B06_135787232 | B06_12479005 | 6.08 | 25.7 |
| WW | VJ | 2013 | qqSCMR.2 | A07 | 150 | A07_61889239 | A07_68857206 | B07 | 140 | B07_1924101 | B07_31546304 | 5.41 | 23.9 | |
| WW | VJ | 2013 | qqSCMR.3 | A03 | 130 | A03_116829847 | A03_16185359 | B03 | 60 | B03_18642606 | B03_13454528 | 5.97 | 20.8 | |
| WW | VJ | 2013 | qqSCMR.4 | A06 | 125 | A06_92969207 | A06_97639398 | B06 | 110 | B06_112293470 | B06_123620597 | 7.9 | 20.0 | |
| WW | VJ | 2013 | qqSCMR.5 | A06 | 60 | A06_70533409 | A06_80763443 | B06 | 65 | B06_87676700 | B06_62922699 | 5.11 | 19.1 | |
| SCMR-ID at 90DAS | WW | VJ | 2013 | qqSCMR.30 | A03 | 140 | A03_121816921 | A03_7178082 | B03 | 70 | B03_135542931 | B03_11838056 | 6.28 | 49.7 |
| WW | VJ | 2013 | qqSCMR.47 | A03 | 130 | A03_116829847 | A03_16185359 | B03 | 60 | B03_18642606 | B03_13454528 | 5.09 | 29.2 | |
| VCR at 30DAS | WW | VJ | 2014 | qqVCR.3 | B05 | 70 | B05_122054913 | B05_148647711 | B07 | 90 | B07_995898 | B07_578122 | 5.75 | 62.9 |
| WW | VJ | 2014 | qqVCR.4 | B01 | 120 | B01_133569092 | B01_3669866 | B03 | 80 | B03_29375836 | B03_5906274 | 5.3 | 32.4 | |
| WW | VJ | 2014 | qqVCR.5 | A01 | 20 | A01_65424740 | A01_105900135 | B01 | 95 | IPAHM569 | B01_114422004 | 5.02 | 15.7 | |
| WW | VJ | 2014 | qqVCR.6 | A06 | 125 | A06_92969207 | A06_97639398 | B06 | 110 | B06_112293470 | B06_123620597 | 5.42 | 14.6 | |
| Traits | QTL Name | Gene Location | Gene Model | Nearest SNP (bp) | Functional Annotation |
|---|---|---|---|---|---|
| A. Drought Tolerance-Related Traits | |||||
| Haulm weight | qHW-B09.1 | Araip.B09 | Araip.1IW39 | 145215085 | MADS-box transcription factor |
| Araip.B09 | Araip.4E9NM | 145215085 | Glycosyl hydrolase | ||
| Araip.B09 | Araip.RXS5A | 16205676 | Malate dehydrogenase | ||
| Araip.B09 | Araip.90KYQ | 16205676 | LOB domain | ||
| Araip.B09 | Araip.UK5PE | 145215085 | Trehalose phosphate synthase | ||
| Araip.B09 | Araip.U4R4L | 16205676 | Protein phosphatase 2C | ||
| Araip.B09 | Araip.KY5AZ | 16205676 | MATE efflux family | ||
| Araip.B09 | Araip.AXK3N | 145215085 | Ethylene-responsive transcription factor | ||
| Pod weight Seed weight | qPW-A03.1 qPW-A03.2 qSW-A03.1 | Aradu.A03 | Aradu.YPV42 | 25161497 | bHLH transcription factor |
| Aradu.A03 | Aradu.TW8M6 | 101625507 | Late embryogenesis abundant (LEA) protein | ||
| Aradu.A03 | Aradu.XX57T | 101625507 | Microtubule-associated protein | ||
| B. Iron Deficiency Tolerance-Related Traits | |||||
| SCMR VCR | qSCMR-ID-B03.1 qSCMR-ID-B03.2 | Araip.B03 | Araip.PW8UQ | 10796590 | NAC domain |
| Araip.B03 | Araip.8EV61 | 13454528 | LRR receptor | ||
| Araip.B03 | Araip.5YM5M | 13454528 | dnaJ-related chaperone protein | ||
| Araip.B03 | Araip.K65JZ | 10796590 | PIP2-5 aquaporin | ||
| Araip.B03 | Araip.VH7YZ | 13454528 | ROP guanine nucleotide exchange factor | ||
| qVCR-B03.1 | Araip.B03 | Araip.IJN8L | 10796590 | 2-Oxoglutarate/Fe(II)-dependent dioxygenase | |
| Araip.B03 | Araip.E5CWX | 13454528 | MYB transcription factor | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, M.K.; Gangurde, S.S.; Sharma, V.; Pattanashetti, S.K.; Naidu, G.K.; Faye, I.; Hamidou, F.; Desmae, H.; Kane, N.A.; Yuan, M.; et al. Improved Genetic Map Identified Major QTLs for Drought Tolerance- and Iron Deficiency Tolerance-Related Traits in Groundnut. Genes 2021, 12, 37. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010037
Pandey MK, Gangurde SS, Sharma V, Pattanashetti SK, Naidu GK, Faye I, Hamidou F, Desmae H, Kane NA, Yuan M, et al. Improved Genetic Map Identified Major QTLs for Drought Tolerance- and Iron Deficiency Tolerance-Related Traits in Groundnut. Genes. 2021; 12(1):37. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010037
Chicago/Turabian StylePandey, Manish K., Sunil S. Gangurde, Vinay Sharma, Santosh K. Pattanashetti, Gopalakrishna K. Naidu, Issa Faye, Falalou Hamidou, Haile Desmae, Ndjido Ardo Kane, Mei Yuan, and et al. 2021. "Improved Genetic Map Identified Major QTLs for Drought Tolerance- and Iron Deficiency Tolerance-Related Traits in Groundnut" Genes 12, no. 1: 37. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010037