
Congruent Genetic and Demographic Dispersal Rates in a Natural Metapopulation at Equilibrium


Abstract
:1. Introduction
- (i)
- determine the genetic structure within a B. eunomia metapopulation based on genetic material collected on more than 1000 individuals over three generations across nine local populations;
- (ii)
- describe the dynamics of the whole-metapopulation through a thorough comparison between genetic and demographic estimates of dispersal rates;
- (iii)
- evaluate the long-term demographic stability of the metapopulation.
2. Materials and Methods
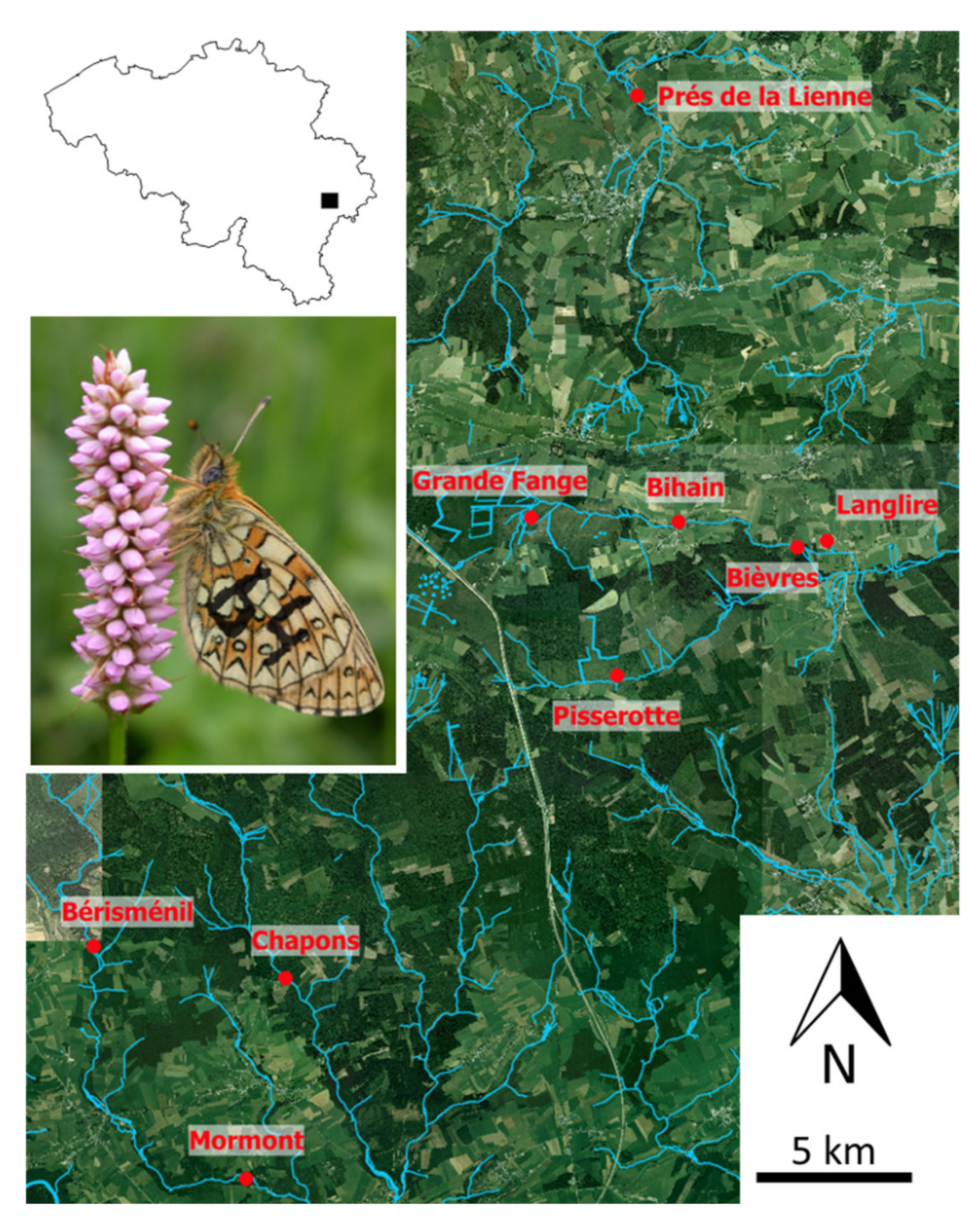
2.1. Model Species and Study Area
2.2. Demographic Modelling: Population Size, Survival and Dispersal from CMR Campaigns
2.3. Molecular Markers, Genetic Structure and Effective Dispersal
2.3.1. Laboratory Work
2.3.2. Population Structure
2.3.3. Isolation by Distance
2.3.4. Allelic Diversity and Effective Population Sizes
2.3.5. Effective Dispersal and Statistical Comparison with Demographic Dispersal
2.3.6. Past Demographic Events
3. Results
3.1. Demography from CMR Data
3.2. Genetic Structure, Isolation by Distance and Diversity
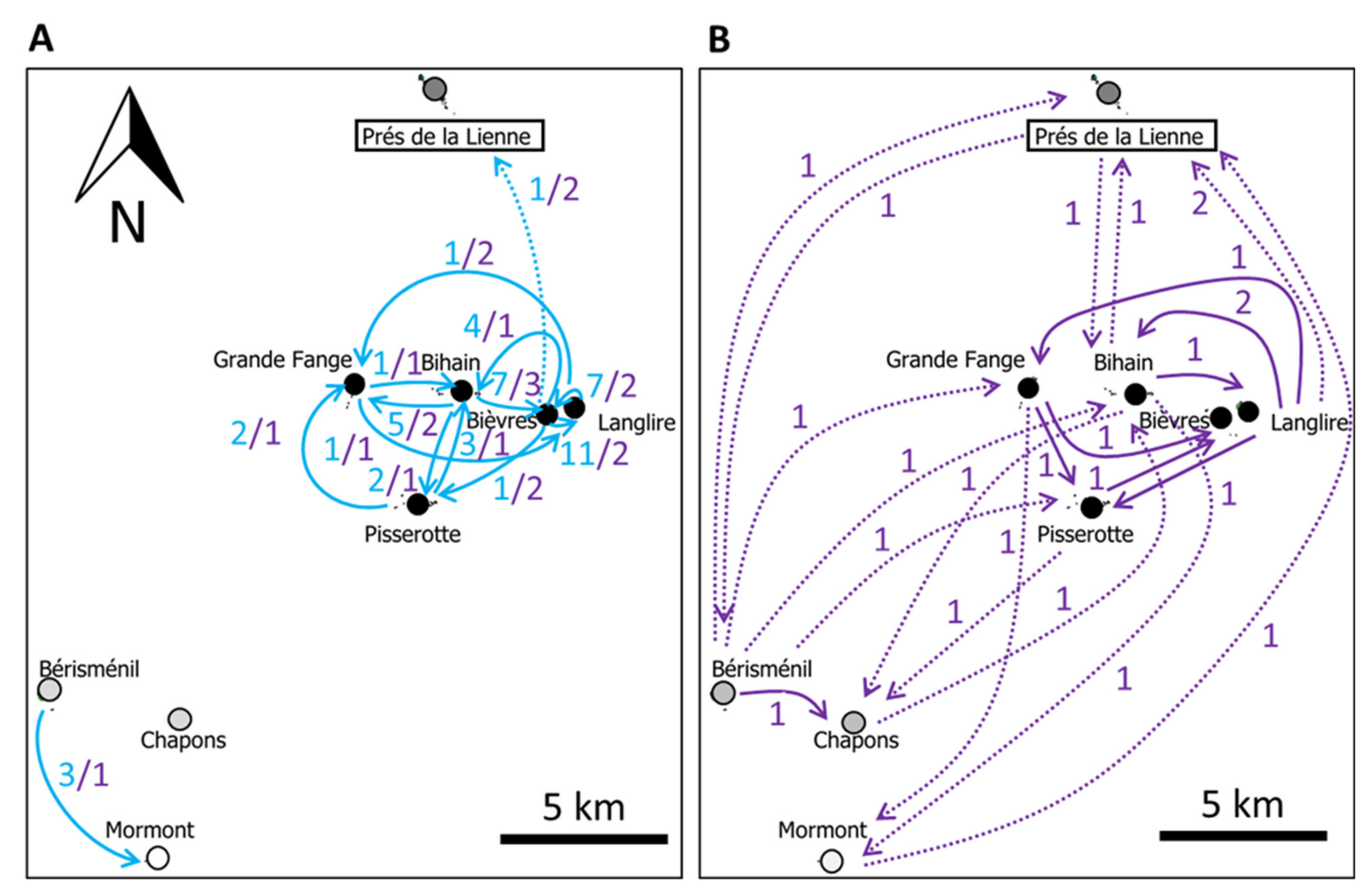
3.3. Comparison between Estimates of Population Sizes and Dispersal
3.4. Past Demographic Events
4. Discussion
4.1. Metapopulation Functioning: An Integrative Story in B. eunomia over Space and Time
4.2. Eco-Evolutionary Perspectives: Controlled Ecological Fluctuations Lead to Long-Term Equilibrium at the Metapopulation Scale
4.3. Methodological Perspectives: Congruence between Demographic and Genetic Estimates of Dispersal
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akçakaya, H.R.; Mills, G.; Doncaster, C.P. The role of metapopulations in conservation. In Key Topics in Conservation Biology; Macdonald, D., Service, K., Eds.; Blackwell Publishing: Oxford, UK, 2007; pp. 64–84. [Google Scholar]
- Levins, R. Extinction. In Some Mathematical Problems in Biology; Gesternhaber, M., Ed.; American Mathematical Soc.: Providence, RI, USA, 1970; pp. 77–107. [Google Scholar]
- Levins, R. Some Demographic and Genetic Consequences of Environmental Heterogeneity for Biological Control. Bull. Entomol. Soc. Am. 1969, 15, 237–240. [Google Scholar] [CrossRef]
- Harrison, S. Local Extinction in a Metapopulation Context: An Empirical Evaluation. Biol. J. Linn. Soc. 1991, 42, 73–88. [Google Scholar] [CrossRef]
- Hanski, I.; Simberloff, D. The Metapopulation Approach, Its History, Conceptual Domain, and Application to Conservation. In Metapopulation Biology, Ecology, Genetics, and Evolution; Hanski, I., Simberloff, D., Eds.; Academic Press: Cambridge, MA, 1997; pp. 5–26. [Google Scholar]
- Kritzer, J.P.; Sale, P.F. Metapopulation Ecology in the Sea: From Levins’ Model to Marine Ecology and Fisheries Science. Fish Fish. 2004, 5, 131–140. [Google Scholar] [CrossRef]
- Baguette, M. The Classical Metapopulation Theory and the Real, Natural World: A Critical Appraisal. Basic Appl. Ecol. 2004, 5, 213–224. [Google Scholar] [CrossRef]
- Fronhofer, E.A.; Kubisch, A.; Hilker, F.M.; Hovestadt, T.; Poethke, H.J. Why Are Metapopulations so Rare? Ecology 2012, 93, 1967–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronce, O. How Does It Feel to Be Like a Rolling Stone? Ten Questions About Dispersal Evolution. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 231–253. [Google Scholar] [CrossRef]
- Clobert, J.; Baguette, M.; Benton, T.G.; Bullock, J.M. Dispersal Ecology and Evolution; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Hanski, I. Habitat Connectivity, Habitat Continuity, and Metapopulations in Dynamic Landscapes. Oikos 1999, 87, 209. [Google Scholar] [CrossRef]
- Baguette, M.; Blanchet, S.; Legrand, D.; Stevens, V.M.; Turlure, C. Individual Dispersal, Landscape Connectivity and Ecological Networks: Dispersal, Connectivity and Networks. Biol. Rev. 2013, 88, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Lenormand, T. Gene Flow and the Limits to Natural Selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- Jacob, S.; Legrand, D.; Chaine, A.S.; Bonte, D.; Schtickzelle, N.; Huet, M.; Clobert, J. Gene Flow Favours Local Adaptation under Habitat Choice in Ciliate Microcosms. Nat. Ecol. Evol. 2017, 1, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Kendall, B.E.; Bjørnstad, O.N.; Bascompte, J.; Keitt, T.H.; Fagan, W.F. Dispersal, Environmental Correlation, and Spatial Synchrony in Population Dynamics. Am. Nat. 2000, 155, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, R.A.; Szűcs, M.; Kasyon, E.; Youngberg, C.; Koontz, M.J.; Richards, C.; Tuff, T.; Melbourne, B.A. Three Types of Rescue Can Avert Extinction in a Changing Environment. Proc. Natl. Acad. Sci. USA 2015, 112, 10557–10562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Haegeman, B.; Loreau, M. Dispersal and Metapopulation Stability. PeerJ 2015, 3, e1295. [Google Scholar] [CrossRef] [PubMed]
- Bohrer, G.; Nathan, R.; Volis, S. Effects of Long-Distance Dispersal for Metapopulation Survival and Genetic Structure at Ecological Time and Spatial Scales. J. Ecol. 2005, 93, 1029–1040. [Google Scholar] [CrossRef]
- Perry, G.L.W.; Lee, F. How Does Temporal Variation in Habitat Connectivity Influence Metapopulation Dynamics? Oikos 2019, 128, 1277–1286. [Google Scholar] [CrossRef]
- Cain, M.L.; Milligan, B.G.; Strand, A.E. Long-Distance Seed Dispersal in Plant Populations. Am. J. Bot. 2000, 87, 1217–1227. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.L.; Loxdale, H.D.; Woiwod, I.P. Monitoring Insect Dispersal: Methods and Approaches. In Dispersal Ecology; Blackwell Science: Oxford, UK, 2002; pp. 24–49. [Google Scholar]
- Katzner, T.E.; Arlettaz, R. Evaluating Contributions of Recent Tracking-Based Animal Movement Ecology to Conservation Management. Front. Ecol. Evol. 2020, 7, 519. [Google Scholar] [CrossRef] [Green Version]
- Lebreton, J.-D.; Burnham, K.P.; Clobert, J.; Anderson, D.R. Modeling Survival and Testing Biological Hypotheses Using Marked Animals: A Unified Approach with Case Studies. Ecol. Monogr. 1992, 62, 67–118. [Google Scholar] [CrossRef] [Green Version]
- Berry, O.; Tocher, M.D.; Sarre, S.D. Can Assignment Tests Measure Dispersal? Mol. Ecol. 2004, 13, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Griesser, M.; Halvarsson, P.; Sahlman, T.; Ekman, J. What Are the Strengths and Limitations of Direct and Indirect Assessment of Dispersal? Insights from a Long-Term Field Study in a Group-Living Bird Species. Behav. Ecol. Sociobiol. 2014, 68, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Chadœuf, J.; Millon, A.; Bourrioux, J.; Printemps, T.; Hecke, B.; Lecoustre, V.; Bretagnolle, V. Modelling Unbiased Dispersal Kernels over Continuous Space by Accounting for Spatial Heterogeneity in Marking and Observation Efforts. Methods Ecol. Evol. 2018, 9, 331–339. [Google Scholar] [CrossRef]
- Clobert, J.; Ims, R.; Rousset, F. Causes, mechanisms and consequences of dispersal. In Ecology, Genetics and Evolution of Metapopulation; Hanski, I., Gaggiotti, O.E., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2004; pp. 307–335. [Google Scholar]
- Schtickzelle, N.; WallisDeVries, M.F.; Baguette, M. Using Surrogate Data in Population Viability Analysis: The Case of the Critically Endangered Cranberry Fritillary Butterfly. Oikos 2005, 109, 89–100. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Whitlock, M.C.; Mccauley, D.E. Indirect Measures of Gene Flow and Migration: FST≠1/(4Nm+1). Heredity 1999, 82, 117–125. [Google Scholar] [CrossRef]
- Prunier, J.G.; Dubut, V.; Chikhi, L.; Blanchet, S. Contribution of Spatial Heterogeneity in Effective Population Sizes to the Variance in Pairwise Measures of Genetic Differentiation. Methods Ecol. Evol. 2017, 8, 1866–1877. [Google Scholar] [CrossRef]
- Broquet, T.; Petit, E.J. Molecular Estimation of Dispersal for Ecology and Population Genetics. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 193–216. [Google Scholar] [CrossRef]
- Cayuela, H.; Rougemont, Q.; Prunier, J.G.; Moore, J.-S.; Clobert, J.; Besnard, A.; Bernatchez, L. Demographic and Genetic Approaches to Study Dispersal in Wild Animal Populations: A Methodological Review. Mol. Ecol. 2018, 27, 3976–4010. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.D.; Epperson, B.K.; Fortin, M.-J.; Holderegger, R.; James, P.M.A.; Rosenberg, M.S.; Scribner, K.T.; Spear, S. Considering Spatial and Temporal Scale in Landscape-Genetic Studies of Gene Flow: Scale in landscape genetics. Mol. Ecol. 2010, 19, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Piry, S.; Alapetite, A.; Cornuet, J.-M.; Paetkau, D.; Baudouin, L.; Estoup, A. GENECLASS2: A Software for Genetic Assignment and First-Generation Migrant Detection. J. Hered. 2004, 95, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.G.; Small, C.M.; Paczolt, K.A.; Ratterman, N.L. A Practical Guide to Methods of Parentage Analysis: TECHNICAL REVIEW. Mol. Ecol. Resour. 2010, 10, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Lowe, W.H.; Allendorf, F.W. What Can Genetics Tell Us about Population Connectivity?: GENETIC AND DEMOGRAPHIC CONNECTIVITY. Mol. Ecol. 2010, 19, 3038–3051. [Google Scholar] [CrossRef]
- Vandewoestijne, S.; Baguette, M. Demographic versus Genetic Dispersal Measures. Popul. Ecol. 2004, 46, 281–285. [Google Scholar] [CrossRef]
- Turlure, C.; Van Dyck, H.; Schtickzelle, N.; Baguette, M. Resource-Based Habitat Definition, Niche Overlap and Conservation of Two Sympatric Glacial Relict Butterflies. Oikos 2009, 118, 950–960. [Google Scholar] [CrossRef]
- Baguette, M.; Vansteenwegen, C.; Convi, I.; Nève, G. Sex-Biased Density-Dependent Migration in a Metapopulation of the Butterfly Proclossiana eunomia. Acta Oecol. 1998, 19, 17–24. [Google Scholar] [CrossRef]
- Baguette, M.; Schtickzelle, N. Negative Relationship between Dispersal Distance and Demography in Butterfly Metapopulations. Ecology 2006, 87, 648–654. [Google Scholar] [CrossRef]
- Turlure, C.; Schtickzelle, N.; Van Dyck, H.; Seymoure, B.; Rutowski, R. Flight Morphology, Compound Eye Structure and Dispersal in the Bog and the Cranberry Fritillary Butterflies: An Inter- and Intraspecific Comparison. PLoS ONE 2016, 11, e0158073. [Google Scholar] [CrossRef]
- Schtickzelle, N.; Baguette, M. Behavioural Responses to Habitat Patch Boundaries Restrict Dispersal and Generate Emigration-Patch Area Relationships in Fragmented Landscapes: Restricted Dispersal and Fragmentation. J. Anim. Ecol. 2003, 72, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Schtickzelle, N.; Le Boulengé, E.; Baguette, M. Metapopulation Dynamics of the Bog Fritillary Butterfly: Demographic Processes in a Patchy Population. Oikos 2002, 97, 349–360. [Google Scholar] [CrossRef]
- Schtickzelle, N.; Baguette, M. Metapopulation Viability Analysis of the Bog Fritillary Butterfly Using RAMAS/GIS. Oikos 2004, 104, 277–290. [Google Scholar] [CrossRef]
- Schtickzelle, N.; Mennechez, G.; Baguette, M. Dispersal Depression with Habitat Fragmentation in the Bog Fritillary Butterfly. Ecology 2006, 87, 1057–1065. [Google Scholar] [CrossRef]
- Baguette, M.; Clobert, J.; Schtickzelle, N. Metapopulation Dynamics of the Bog Fritillary Butterfly: Experimental Changes in Habitat Quality Induced Negative Density-Dependent Dispersal. Ecography 2011, 34, 170–176. [Google Scholar] [CrossRef]
- Nève, G.; Barascud, B.; Descimon, H.; Baguette, M. Genetic Structure of Proclossiana eunomia Populations at the Regional Scale (Lepidoptera, Nymphalidae). Heredity 2000, 84, 657–666. [Google Scholar] [CrossRef]
- Nève, G.; Mousson, L.; Baguette, M. Adult Dispersal and Genetic Structure of Butterfly Populations in a Fragmented Landscape. Acta Oecol. 1996, 17, 621–626. [Google Scholar]
- Maresova, J.; Habel, J.C.; Neve, G.; Sielezniew, M.; Bartonova, A.; Kostro-Ambroziak, A.; Fric, Z.F. Cross-Continental Phylogeography of Two Holarctic Nymphalid Butterflies, Boloria eunomia and Boloria Selene. PLoS ONE 2019, 14, e0214483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewoestijne, S.; Baguette, M. Genetic Population Structure of the Vulnerable Bog Fritillary Butterfly: Genetic Population Structure of the Bog Fritillary Butterfly. Hereditas 2005, 141, 199–206. [Google Scholar] [CrossRef] [PubMed]
- White, G.C.; Burnham, K.P. Program MARK: Survival Estimation from Populations of Marked Animals. Bird Study 1999, 46, S120–S139. [Google Scholar] [CrossRef]
- Cooch, E.; White, G.C. Program Mark: A Gentle Introduction; Colorado State University: Fort Collins, CO, USA, 2002; Available online: http://www.phidot.org/software/mark/docs/book/.
- Legrand, D.; Chaput-Bardy, A.; Turlure, C.; Dubois, Q.; Huet, M.; Schtickzelle, N.; Stevens, V.M.; Baguette, M. Isolation and Characterization of 15 Microsatellite Loci in the Specialist Butterfly Boloria eunomia. Conserv. Genet. Resour. 2014, 6, 223–227. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Narum, S.R. Beyond Bonferroni: Less Conservative Analyses for Conservation Genetics. Conserv. Genet. 2006, 7, 783–787. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Coulon, A.; Morellet, N.; Goulard, M.; Cargnelutti, B.; Angibault, J.-M.; Hewison, A.J.M. Inferring the Effects of Landscape Structure on Roe Deer (Capreolus Capreolus) Movements Using a Step Selection Function. Landsc. Ecol. 2008, 23, 603–614. [Google Scholar] [CrossRef]
- Rousset, F. Genetic Differentiation and Estimation of Gene Flow from FStatistics Under Isolation by Distance. Genet 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- Glaubitz, J.C. Convert: A User-Friendly Program to Reformat Diploid Genotypic Data for Commonly Used Population Genetic Software Packages. Mol. Ecol. Notes 2004, 4, 309–310. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. Linkage Disequilibrium Estimates of Contemporary N e Using Highly Variable Genetic Markers: A Largely Untapped Resource for Applied Conservation and Evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-Implementation of Software for the Estimation of Contemporary Effective Population Size (Ne) from Genetic Data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Rannala, B.; Mountain, J.L. Detecting Immigration by Using Multilocus Genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef] [Green Version]
- Paetkau, D.; Slade, R.; Burden, M.; Estoup, A. Genetic Assignment Methods for the Direct, Real-Time Estimation of Migration Rate: A Simulation-Based Exploration of Accuracy and Power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.-M.; Luikart, G. Description and Power Analysis of Two Tests for Detecting Recent Population Bottlenecks From Allele Frequency Data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Luikart, G. Distortion of Allele Frequency Distributions Provides a Test for Recent Population Bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Thrall, P.H.; Burdon, J.J.; Murray, B.R. The metapopulation paradigm: A fragmented view of conservation biology. In Genetics, Demography and Viability of Fragmented Populations; Young, A.G., Clarke, G.M., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 75–96. [Google Scholar]
- Frankham, R.; Bradshaw, C.J.A.; Brook, B.W. Genetics in Conservation Management: Revised Recommendations for the 50/500 Rules, Red List Criteria and Population Viability Analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]
- Maggs, C.A.; Castilho, R.; Foltz, D.; Henzler, C.; Jolly, M.T.; Kelly, J.; Olsen, J.; Perez, K.E.; Stam, W.; La, R.V.I. Evaluating signatures of glacial refugia for north atlantic benthic marine taxa. Ecology 2008, 89, S108–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennington, W.J.; Gockel, J.; Partridge, L. Testing for Asymmetrical Gene Flow in a Drosophila melanogaster Body-Size Cline. Genetics 2003, 165, 667–673. [Google Scholar] [PubMed]
- Watts, P.C.; Rousset, F.; Saccheri, I.J.; Leblois, R.; Kemp, S.J.; Thompson, D.J. Compatible Genetic and Ecological Estimates of Dispersal Rates in Insect (Coenagrion Mercuriale: Odonata: Zygoptera) Populations: Analysis of ‘Neighbourhood Size’ Using a More Precise Estimator: Neighbourhood size in coenagrion mercuriale. Mol. Ecol. 2006, 16, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Dias, P.C. Sources and Sinks in Population Biology. Trends Ecol. Evol. 1996, 11, 326–330. [Google Scholar] [CrossRef]
- Barraquand, F.; Louca, S.; Abbott, K.C.; Cobbold, C.A.; Cordoleani, F.; DeAngelis, D.L.; Elderd, B.D.; Fox, J.W.; Greenwood, P.; Hilker, F.M.; et al. Moving Forward in Circles: Challenges and Opportunities in Modelling Population Cycles. Ecol. Lett. 2017, 20, 1074–1092. [Google Scholar] [CrossRef]
- Choutt, J.; Turlure, C.; Baguette, M.; Schtickzelle, N. Parasitism Cost of Living in a High Quality Habitat in the Bog Fritillary Butterfly. Biodivers Conserv. 2011, 20, 3117–3131. [Google Scholar] [CrossRef]
- Choutt, J. The Butterfly and the Wasp: Host Parasitoid Relationship between Boloria Eunomia and Cotesia Eunomiae; UCLouvain: Louvain-la-Neuve, Belgium, 2011. [Google Scholar]
- Turlure, C. Habitat from a Butterfly’s Point of View: How Specialist Butterflies Map onto Ecological Resources; UCLouvain: Louvain-la-Neuve, Belgium, 2009. [Google Scholar]
- Turlure, C.; Legrand, D.; Schtickzelle, N.; Baguette, M. Male Disguised Females: Costs and Benefits of Female-Limited Dimorphism in a Butterfly: Evolutionary Ecology of Female-Limited Dimorphism. Ecol. Entomol. 2016, 41, 572–581. [Google Scholar] [CrossRef]
- Yaari, G.; Ben-Zion, Y.; Shnerb, N.M.; Vasseur, D.A. Consistent Scaling of Persistence Time in Metapopulations. Ecology 2012, 93, 1214–1227. [Google Scholar] [CrossRef]
- Burgess, S.C.; Baskett, M.L.; Grosberg, R.K.; Morgan, S.G.; Strathmann, R.R. When Is Dispersal for Dispersal? Unifying Marine and Terrestrial Perspectives: When Is Dispersal for Dispersal? Biol. Rev. 2016, 91, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Turlure, C.; Baguette, M.; Stevens, V.M.; Maes, D. Species- and Sex-Specific Adjustments of Movement Behavior to Landscape Heterogeneity in Butterflies. Behav. Ecol. 2011, 22, 967–975. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 2009 | 2010 | 2011 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Population | N | Ne | CI Ne | N | Ne | CI Ne | N | Ne | CI Ne |
| Bérismenil | 196 | 58.6 | 27.4–410.1 | 191 | 35 | 23.6–54.5 | 235 | 102 | 39.8–∞ |
| Chapons | 14 | NA | NA | 13 | 1.9 | 1.1–6.1 | 8 | 14.6 | 1.7–∞ |
| Mormont | 25 | 4.7 | 1.7–32.2 | 30 | 9.9 | 3.9–24.6 | 56 | 12.8 | 4.4–59.1 |
| Bièvres | 195 | 50.5 | 19.2–∞ | 259 | 44.8 | 26.4–92.5 | 304 | 46 | 26.9–109.9 |
| Bihain | 136 | 22.4 | 12.3–54.3 | 419 | 64.4 | 40.6–118.5 | 289 | 111 | 52.4–990.9 |
| Grande Fange | 70 | 17 | 3–∞ | 166 | 55.8 | 26.8–240.2 | 336 | 113 | 34–∞ |
| Langlire | 57 | 91.2 | 12.5–∞ | 101 | 56.5 | 23–∞ | 99 | 95.3 | 23.8–∞ |
| Pisserotte | 978 | 34.3 | 14.2–522.8 | 1428 | 142.6 | 72.7–657.9 | 1139 | NA | 84.9–∞ |
| Prés de la Lienne | 291 | 46.4 | 26.8–105.1 | 940 | 57.7 | 24.6–∞ | 1131 | 122 | 75.4–241.1 |
| Population | Year | n | Na | Np | A | Ap | Ho | He | FIS | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|
| Bersimenil | 2009 | 47 | 5.3 | 2 | 3.54 | 0.18 | 0.44 | 0.64 | 0.17 | 0.0003 |
| 2010 | 97 | 5.1 | 0 | 3.69 | 0.16 | 0.47 | 0.61 | 0.08 | 0.002 | |
| 2011 | 56 | 5.2 | 0 | 3.64 | 0.11 | 0.52 | 0.67 | 0.09 | 0.004 | |
| Chapons | 2009 | 6 | 2.9 | 0 | 2.5 | 0.09 | 0.6 | 0.61 | −0.15 | 0.6 |
| 2010 | 11 | 3 | 0 | 2.69 | 0.05 | 0.41 | 0.45 | −0.01 | 0.59 | |
| 2011 | 9 | 3.5 | 1 | 3.13 | 0.14 | 0.43 | 0.61 | 0.24 | 0.01 | |
| Mormont | 2009 | 13 | 3 | 2 | 2.74 | 0.27 | 0.46 | 0.57 | 0.008 | 0.51 |
| 2010 | 22 | 3.6 | 0 | 2.94 | 0.18 | 0.34 | 0.58 | 0.26 | <0.0001 | |
| 2011 | 24 | 3.6 | 0 | 2.84 | 0.17 | 0.39 | 0.55 | 0.16 | 0.006 | |
| Bièvres | 2009 | 36 | 5.6 | 1 | 3.74 | 0.12 | 0.43 | 0.68 | 0.18 | <0.0001 |
| 2010 | 67 | 6.4 | 2 | 3.91 | 0.15 | 0.48 | 0.68 | 0.15 | <0.0001 | |
| 2011 | 38 | 6.1 | 2 | 3.8 | 0.15 | 0.48 | 0.69 | 0.13 | 0.0003 | |
| Bihain | 2009 | 31 | 5.6 | 2 | 3.79 | 0.21 | 0.46 | 0.69 | 0.18 | <0.0001 |
| 2010 | 99 | 6.7 | 2 | 3.87 | 0.18 | 0.47 | 0.68 | 0.13 | <0.0001 | |
| 2011 | 63 | 6.4 | 3 | 3.81 | 0.18 | 0.48 | 0.7 | 0.15 | <0.0001 | |
| Grande Fange | 2009 | 15 | 4.4 | 4 | 3.52 | 0.4 | 0.49 | 0.64 | 0.13 | 0.47 |
| 2010 | 57 | 6.5 | 5 | 4.06 | 0.21 | 0.48 | 0.69 | 0.17 | <0.0001 | |
| 2011 | 34 | 5.8 | 4 | 3.76 | 0.21 | 0.47 | 0.68 | 0.12 | 0.002 | |
| Langlire | 2009 | 17 | 5.1 | 2 | 3.69 | 0.17 | 0.45 | 0.68 | 0.19 | 0.004 |
| 2010 | 42 | 5.4 | 2 | 3.99 | 0.09 | 0.46 | 0.68 | 0.19 | <0.0001 | |
| 2011 | 28 | 5.6 | 0 | 3.75 | 0.09 | 0.41 | 0.68 | 0.18 | <0.0001 | |
| Pisserotte | 2009 | 36 | 5.3 | 2 | 3.56 | 0.06 | 0.47 | 0.67 | 0.09 | 0.03 |
| 2010 | 83 | 6 | 0 | 3.94 | 0.15 | 0.49 | 0.69 | 0.13 | <0.0001 | |
| 2011 | 31 | 5.3 | 0 | 3.74 | 0.04 | 0.49 | 0.69 | 0.15 | 0.0008 | |
| Prés Lienne | 2009 | 57 | 5.1 | 2 | 3.31 | 0.12 | 0.42 | 0.6 | 0.14 | <0.0001 |
| 2010 | 35 | 4.6 | 1 | 3.27 | 0.11 | 0.41 | 0.62 | 0.15 | 0.0003 | |
| 2011 | 163 | 5.4 | 3 | 3.3 | 0.08 | 0.41 | 0.6 | 0.23 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Legrand, D.; Baguette, M.; Prunier, J.G.; Dubois, Q.; Turlure, C.; Schtickzelle, N. Congruent Genetic and Demographic Dispersal Rates in a Natural Metapopulation at Equilibrium. Genes 2021, 12, 362. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030362
Legrand D, Baguette M, Prunier JG, Dubois Q, Turlure C, Schtickzelle N. Congruent Genetic and Demographic Dispersal Rates in a Natural Metapopulation at Equilibrium. Genes. 2021; 12(3):362. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030362
Chicago/Turabian StyleLegrand, Delphine, Michel Baguette, Jérôme G. Prunier, Quentin Dubois, Camille Turlure, and Nicolas Schtickzelle. 2021. "Congruent Genetic and Demographic Dispersal Rates in a Natural Metapopulation at Equilibrium" Genes 12, no. 3: 362. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030362