DEEPGENTM—A Novel Variant Calling Assay for Low Frequency Variants

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
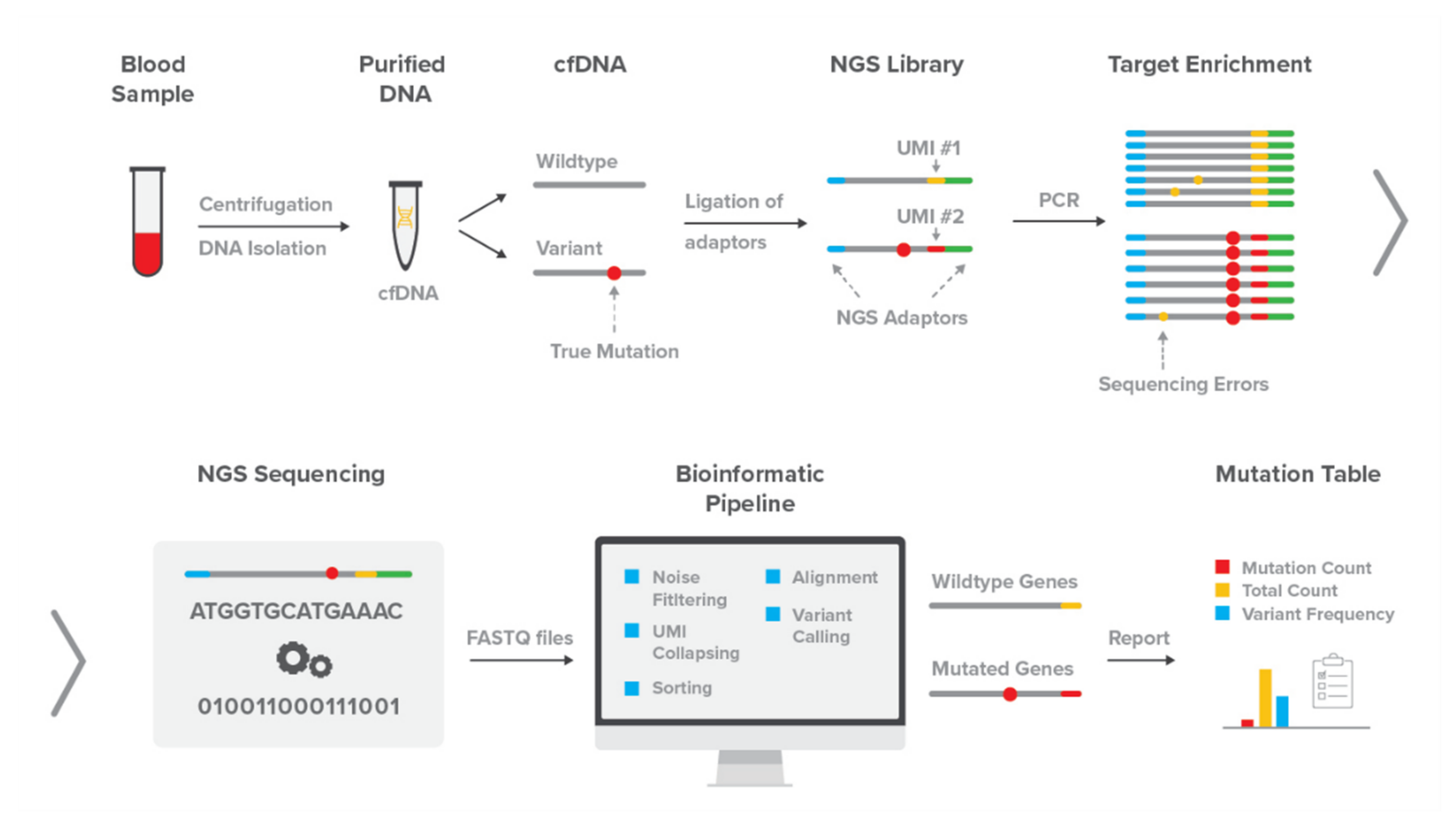
2.1. The DEEPGENTM Assay
2.1.1. Library Preparation and Sequencing
2.1.2. DEEPGENTM Bioinformatics Pipeline
2.2. Assay Validation
2.2.1. Sample Selection
2.2.2. Analytical Validation
2.2.3. Orthogonal Validation
3. Results
3.1. DEEPGENTM Performance Analysis
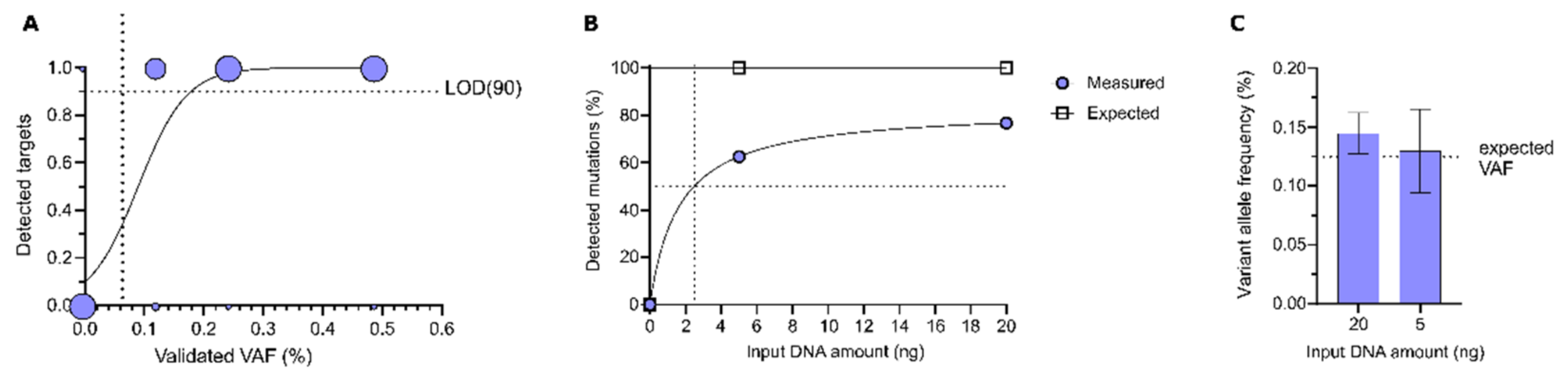
3.1.1. Sensitivity of DEEPGENTM
3.1.2. Intra-Assay Reproducibility
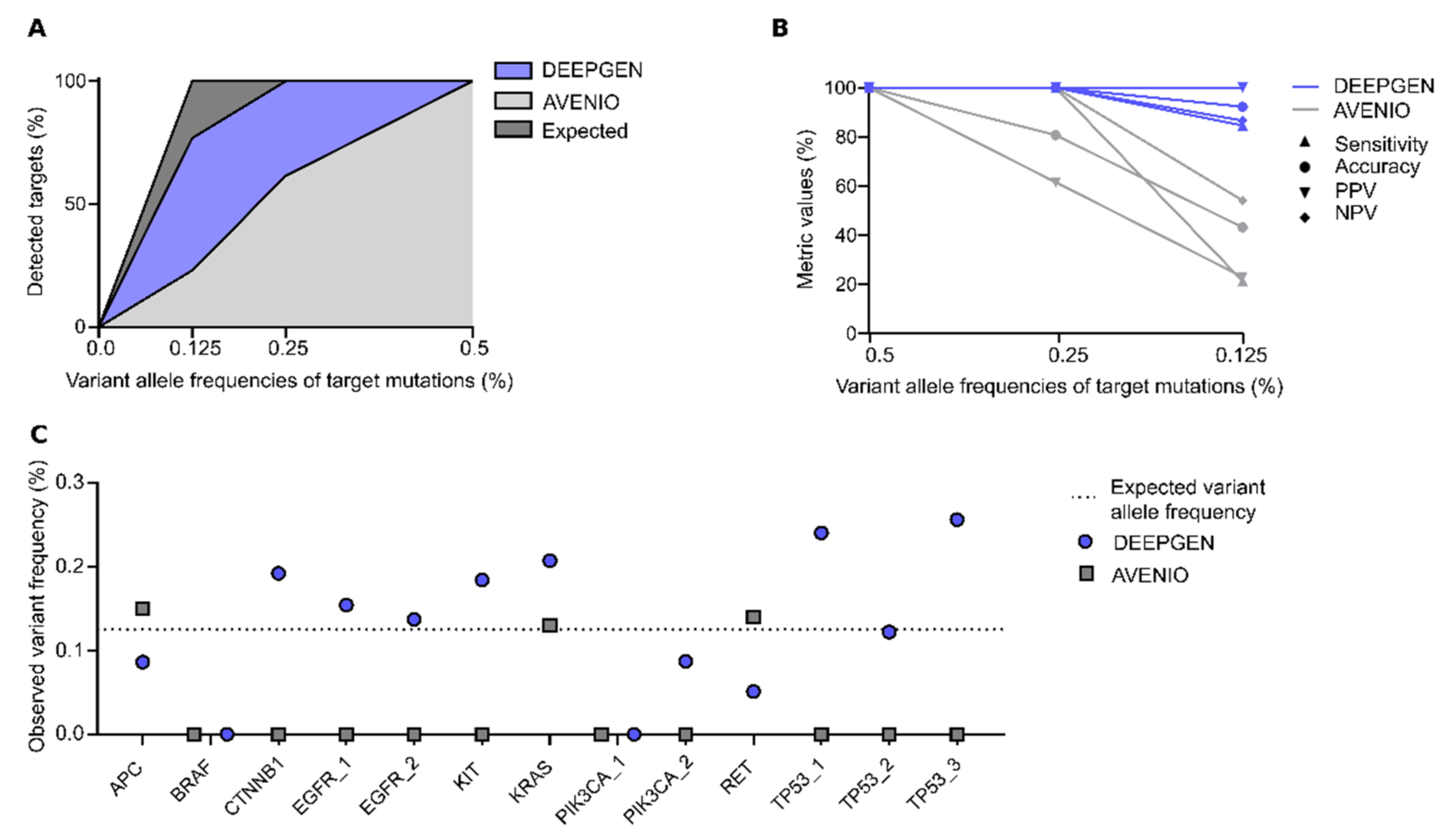
3.2. Orthogonal Assay Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, W.; Li, A.; Kansal, R.; Chen, Y.; Chen, H.; Li, X. Clinical Next Generation Sequencing for Precision Medicine in Cancer. Curr. Genom. 2015, 16, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, B.; Nakaoka, H.; Akhondzadeh, S.; Tekin, M.; Mahdieh, N. Next generation sequencing: Implications in personalized medicine and pharmacogenomics. Mol. BioSyst. 2016, 12, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, R.L.; Priest, J.R.; Zook, J.M.; Grove, M.E.; Waggott, D.; Wheeler, M.T.; Salit, M.; Ashley, E.A. Medical implications of technical accuracy in genome sequencing. Genome Med. 2016, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranneheim, H.; Wedell, A. Exome and genome sequencing: A revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 2016, 279, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Eberle, M.A.; Fritzilas, E.; Krusche, P.; Källberg, M.; Moore, B.L.; Bekritsky, M.A.; Iqbal, Z.; Chuang, H.-Y.; Humphray, S.J.; Halpern, A.L.; et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017, 27, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garraway, L.A.; Verweij, J.; Ballman, K.V. Precision Oncology: An Overview. J. Clin. Oncol. 2013, 31, 1803–1805. [Google Scholar] [CrossRef] [Green Version]
- Robasky, K.; Lewis, N.E.; Church, G.M. The role of replicates for error mitigation in next-generation sequencing. Nat. Rev. Genet. 2014, 15, 56–62. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, D.; Lim, J.S.; Maeng, J.H.; Son, H.; Kang, H.-C.; Nam, H.; Lee, J.H.; Kim, S. The use of technical replication for detection of low-level somatic mutations in next-generation sequencing. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, S.; De Graaf, A.O.; Karimi, M.; Van Der Reijden, B.A.; Hellström-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SoRelle, J.A.; Wachsmann, M.; Cantarel, B.L. Assembling and Validating Bioinformatic Pipelines for Next-Generation Sequencing Clinical Assays. Arch. Pathol. Lab. Med. 2020, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daber, R.; Sukhadia, S.; Morrissette, J.J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet. 2013, 206, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Zhu, B.; Wang, M.; Hu, Y.; Chen, Q.; Nguyen, C.; Hicks, B.; Meerzaman, D. Comparing the performance of selected variant callers using synthetic data and genome segmentation. BMC Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, A.; Golicz, A.; Hackett, C.A.; Milne, I.; Stephen, G.; Marshall, D.; Flavell, A.J.; Bayer, M. An investigation of causes of false positive single nucleotide polymorphisms using simulated reads from a small eukaryote genome. BMC Bioinform. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Dannebaum, R.; Singh, A.; Foley, R.; Dinis, J.; Choi, C.; Min, B.; Li, J.; Feng, L.; Casey, F.; et al. Abstract 3648: Performance of the AVENIO ctDNA assays across multiple high-throughput next-generation sequencing platforms. Cancer Res. 2018, 78, 3648. [Google Scholar] [CrossRef]
- Li, H.; Wren, J. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobo, D.; Lipatov, M.; Rodriguez-Flores, J.L.; Auton, A.; Henn, B.M. False Negatives Are a Significant Feature of Next Generation Sequencing Callsets. bioRxiv 2016, 066043. [Google Scholar] [CrossRef] [Green Version]
- Jung, A.; Kirchner, T. Liquid biopsy in tumor genetic diagnosis. Dtsch. Arztebl. Int. 2018, 115, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance Metrics | Formula | VAF Specific Results | Overall Results | Overall 95% CI | |
|---|---|---|---|---|---|
| Sensitivity | 0.5% 0.25% 0.125% | 98.3% 98.3% 76.7% | 91.1% | 86.0%–94.8% | |
| Specificity | 0% | 95% | 95.0% | 86.1%–99.0% | |
| PPV | 0.5% 0.25% 0.125% | 95.2% 95.2% 93.9% | 98.2% | 94.8%–99.6% | |
| NPV | 0.5% 0.25% 0.125% | 98.3% 98.3% 80.3% | 78.1% | 66.8%–86.9% | |
| Accuracy | 0.5% 0.25% 0.125% | 96.7% 96.7% 85.8% | 92.1% | 87.9%–95.2% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermann, B.T.; Pfeil, S.; Groenke, N.; Schaible, S.; Kunze, R.; Ris, F.; Hagen, M.E.; Bhakdi, J. DEEPGENTM—A Novel Variant Calling Assay for Low Frequency Variants. Genes 2021, 12, 507. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040507
Hermann BT, Pfeil S, Groenke N, Schaible S, Kunze R, Ris F, Hagen ME, Bhakdi J. DEEPGENTM—A Novel Variant Calling Assay for Low Frequency Variants. Genes. 2021; 12(4):507. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040507
Chicago/Turabian StyleHermann, Bernd Timo, Sebastian Pfeil, Nicole Groenke, Samuel Schaible, Robert Kunze, Frédéric Ris, Monika Elisabeth Hagen, and Johannes Bhakdi. 2021. "DEEPGENTM—A Novel Variant Calling Assay for Low Frequency Variants" Genes 12, no. 4: 507. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040507