
Dynamic Expression of Imprinted Genes in the Developing and Postnatal Pituitary Gland

 , , ,
, , ,
Abstract
:1. Introduction
1.1. The Pituitary Gland
1.1.1. Function
1.1.2. Development
1.1.3. Endocrine Aspects of Human Imprinting Disorders
1.1.4. Co-Ordinate Regulation of Imprinted Genes
1.1.5. Broader Roles for Imprinted Genes in Maternal-Offspring Communication
1.2. Aims of the Study
2. Materials and Methods
2.1. Mice
2.2. Embryo vs. Adult Comparison
2.3. Single-Cell RNA Sequencing (scRNAseq) Data Processing
2.4. Pregnant vs. Virgin Comparison
2.5. Histology
2.6. In Situ Hybridisation
2.7. Immunohistochemistry
3. Results
3.1. Imprinted Genes Are Highly Expressed in the Anterior Pituitary Gland and Are Developmentally Regulated
3.2. “Embryo High” Imprinted Gene Expression Is Not Retained in the Postnatal Stem Cell Population

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Combined P4 & P49 | P4 | P49 |
|---|---|---|---|
| Stem Cells | Igf2r, Pdk4, Kcnq1ot1, Zim1, Peg3, Sgce, Plagl1, Zdbf2, Usp29, Ube3a | Gab1, Igf2r, Peg3, Zim1, Kcnq1ot1, Pdk4, Plagl1, Zdbf2 | Pdk4, Pon2, Sgce, Plagl1, Kcnq1ot1 |
| Proliferating Cells | Plagl1, Commd1, Zdbf2, Tssc4, Mdh2 | Tssc4, Mdh2, Commd1, Zdbf2 | Tssc4, Commd1, Mdh2, Zdbf2, Ube3a |
| Somatotrophs | Dlk1 | Dlk1 | |
| Lactotrophs | Meg3, Blcap, H13, Cdkn1c, Nap1l5 | Nnat, Asb4, Meg3, Blcap, H13, Nap1l5 | |
| Thyrotrophs | Peg10, Gnas, Mcts2, Snrpn | Dlk1, Gnas, Peg10, Nap1l5, Snrpn, Mcts2 | |
| Melanotrophs | Usp29 | Usp29 | |
| Corticotrophs | Usp29 | Nap1l5, Usp29, Magi2, Impact | Usp29 |
| Sf1 progenitors | Bcl2l1 | Bcl2l1, H13, Blcap | Cdkn1c |
| Gonadotrophs | Qpct, Rasgrf1, Ndn, Nnat | Qpct, Th, Nnat | Rasgrf1, Qpct, Gnas, Snrpn, Ndn, Kcnq1ot1, Mdh2 |
3.3. Clustered Imprinted Genes Show Overlapping Cell-Specific Gene Expression
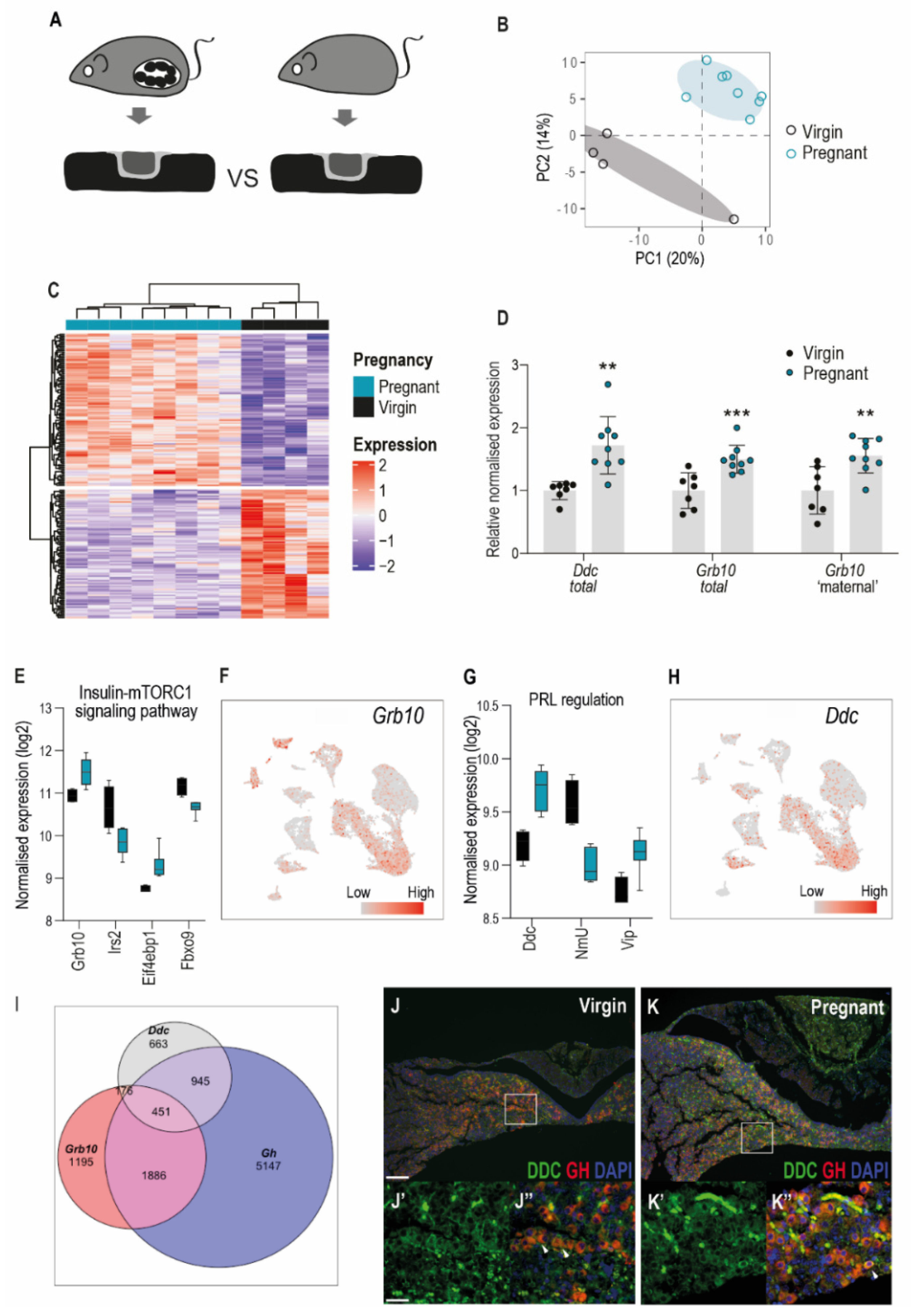
3.4. Imprinted Gene Expression in the Pituitary Gland of the Pregnant Dam
4. Discussion and Conclusions
4.1. Role of Imprinted Genes in Pituitary Development and the Imprinted Gene Network
4.2. Pituitary IG Expression and Relevance to Human Disease
4.3. Imprinted Genes in the Pregnant Pituitary Gland
4.4. Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic imprinting and physiological processes in mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Barton, S.C.; Surani, M.A.H.; Norris, M.L. Role of paternal and maternal genomes in mouse development. Nature 1984, 311, 374–376. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Cleaton, M.A.M.; Edwards, C.A.; Ferguson-Smith, A.C. Phenotypic outcomes of imprinted gene models in mice: Elucidation of pre- and postnatal functions of imprinted genes. Annu. Rev. Genom. Hum. Genet. 2014, 15, 93–126. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.J.; Robinson, D.O.; Lamont, L.; Shield, J.; Temple, I.K. Paternal uniparental disomy of chromosome 6 and transient neonatal diabetes mellitus. Clin. Genet. 2008, 54, 522–525. [Google Scholar] [CrossRef]
- Gardner, R.J. An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 2000, 9, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temple, I.K.; James, R.S.; Crolla, J.A.; Sitch, F.L.; Jacobs, P.A.; Howell, W.M.; Betts, P.; Baum, J.D.; Shield, J.P.H. An imprinted gene(s) for diabetes? Nat. Genet. 1995, 9, 110–112. [Google Scholar] [CrossRef]
- Arima, T.; Drewell, R.A.; Oshimura, M.; Wake, N.; Surani, M. A Novel Imprinted Gene, HYMAI, Is Located within an Imprinted Domain on Human Chromosome 6 Containing ZAC. Genomics 2000, 67, 248–255. [Google Scholar] [CrossRef]
- Kamiya, M.; Judson, H.; Okazaki, Y.; Kusakabe, M.; Muramatsu, M.; Takada, S.; Takagi, N.; Arima, T.; Wake, N.; Kamimura, K.; et al. The cell cycle control gene ZAC/PLAGL1 is imprinted--a strong candidate gene for transient neonatal dia-betes. Hum. Mol. Genet. 2000, 9, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Begemann, M.; Zirn, B.; Santen, G.; Wirthgen, E.; Soellner, L.; Büttel, H.-M.; Schweizer, R.; van Workum, W.; Binder, G.; Eggermann, T. Paternally inherited IGF2 mutation and growth restriction. N. Engl. J. Med. 2015, 373, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotzot, D.; Schmitt, S.; Bernasconi, F.; Robinson, W.P.; Lurie, I.W.; Ilyina, H.; Méhes, K.; Hamel, B.C.; Otten, B.J.; Hergersberg, M.; et al. Uniparental disomy 7 in Silver—Russell syndrome and primordial growth retardation. Hum. Mol. Genet. 1995, 4, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Cytrynbaum, C.; Chong, K.; Hannig, V.; Choufani, S.; Shuman, C.; Steele, L.; Morgan, T.; Scherer, S.W.; Stavropoulos, D.J.; Basran, R.K.; et al. Genomic imbalance in the centromeric 11p15 imprinting center in three families: Further evidence of a role for IC2 as a cause of Russell-Silver syndrome. Am. J. Med. Genet. Part A 2016, 170, 2731–2739. [Google Scholar] [CrossRef]
- Eggermann, T.; Wollmann, H.A.; Kuner, R.; Eggermann, K.; Enders, H.; Kaiser, P.; Ranke, M.B. Molecular studies in 37 Silver-Russell syndrome patients: Frequency and etiology of uniparental disomy. Qual. Life Res. 1997, 100, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Preece, M.A.; Price, S.M.; Davies, V.; Clough, L.; Stanier, P.; Trembath, R.C.; Moore, G.E. Maternal uniparental disomy 7 in Silver-Russell syndrome. J. Med. Genet. 1997, 34, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicquel, C.; Rossignol, S.; Cabrol, S.; Houang, M.; Steunou, V.; Barbu, V.; Danton, F.; Thibaud, N.; Le Merrer, M.; Burglen, L.; et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 2005, 37, 1003–1007. [Google Scholar] [CrossRef]
- Koufos, A.; Grundy, P.; Morgan, K.; Aleck, K.A.; Hadro, T.; Lampkin, B.C.; Kalbakji, A.; Cavenee, W.K. Familial Wiedemann-Beckwith syndrome and a second Wilms tumor locus both map to 11p15.5. Am. J. Hum. Genet. 1989, 44, 711–719. [Google Scholar]
- Ping, A.J.; E Reeve, A.; Law, D.J.; Young, M.R.; Boehnke, M.; Feinberg, A.P. Genetic linkage of Beckwith-Wiedemann syndrome to 11p15. Am. J. Hum. Genet. 1989, 44, 720–723. [Google Scholar]
- Reik, W.; Brown, K.W.; Schneid, H.; Le Bouc, Y.; Bickmore, W.; Maher, E.R. Imprinting mutations in the Beckwith—Wiedemann syndrome suggested by an altered imprinting pattern in the IGF2–H19 domain. Hum. Mol. Genet. 1995, 4, 2379–2385. [Google Scholar] [CrossRef]
- Brown, K.W.; Villar, A.J.; Bickmore, W.; Clayton-Smith, J.; Catchpoole, D.; Maher, E.R.; Reik, W. Imprinting mutation in the Beckwith-Wiedemann syndrome leads to biallelic IGF2 expression through an H19-independent pathway. Hum. Mol. Genet. 1996, 5, 2027–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demars, J.; Shmela, M.E.; Rossignol, S.; Okabe, J.; Netchine, I.; Azzi, S.; Cabrol, S.; Le Caignec, C.; David, A.; Le Bouc, Y.; et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum. Mol. Genet. 2009, 19, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syn-drome. Nat. Genet. 2004, 36, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Hatada, I.; Ohashi, H.; Fukushima, Y.; Kaneko, Y.; Inoue, M.; Komoto, Y.; Okada, A.; Ohishi, S.; Nabetani, A.; Morisaki, H.; et al. An imprinted gene p57KIP2 is mutated in Beckwith–Wiedemann syndrome. Nat. Genet. 1996, 14, 171–173. [Google Scholar] [CrossRef]
- Brioude, F.; Netchine, I.; Praz, F.; Le Jule, M.; Calmel, C.; Lacombe, D.; Edery, P.; Catala, M.; Odent, S.; Isidor, B.; et al. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Hum. Mutat. 2015, 36, 894–902. [Google Scholar] [CrossRef]
- Arboleda, V.A.; Lee, H.; Parnaik, R.; Fleming, A.; Banerjee, A.; Ferraz-de-Souza, B.; Délot, E.C.; Rodriguez-Fernandez, I.A.; Braslavsky, D.; Bergadá, I.; et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet. 2012, 44, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Tommiska, J.; Känsäkoski, J.; Skibsbye, L.; Vaaralahti, K.; Liu, X.; Lodge, E.J.; Tang, C.; Yuan, L.; Fagerholm, R.; Kanters, J.K.; et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat. Commun. 2017, 8, 1289. [Google Scholar] [CrossRef] [Green Version]
- Temple, I.K.; Cockwell, A.; Hassold, T.; Pettay, D.; Jacobs, P. Maternal uniparental disomy for chromosome 14. J. Med. Genet. 1991, 28, 511–514. [Google Scholar] [CrossRef]
- Ogata, T.; Kagami, M. Kagami–Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 2016, 61, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Dauber, A.; Cunha-Silva, M.; Macedo, D.B.; Brito, V.N.; Abreu, A.P.; Roberts, S.A.; Montenegro, L.R.; Andrew, M.; Kirby, A.; Weirauch, M.T.; et al. Paternally inherited DLK1 deletion associated with familial central precocious puberty. J. Clin. Endocrinol. Metab. 2017, 102, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Burman, P.; Ritzen, E.M.; Lindgren, A.C. Endocrine dysfunction in Prader-Willi syndrome: A review with special reference to GH. Endocr. Rev. 2001, 22, 787–799. [Google Scholar] [CrossRef]
- Driscoll, D.J.; Miller, J.M.; Schwartz, S.; Cassidy, S.B. Prader-Willi syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Butler, M.G.; Palmer, C.G. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet 1983, 1, 1285–1286. [Google Scholar] [CrossRef]
- Sahoo, T.; Del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.D.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef] [Green Version]
- De Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Sutcliffe, J.S.; Fang, P.; Galjaard, R.-J.; Jiang, Y.-H.; Benton, C.S.; Rommens, J.M.; Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997, 15, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, L.C.; Shah, P.; Sanner, J.R.F.; Arancibia, M.; Hurst, J.; Jones, W.; Spoudeas, H.; le Quesne Stabej, P.; Williams, H.J.; Ocaka, L.A.; et al. Mutations in MAGEL2 and L1CAM are associated with congenital hypopituitarism and arthrogryposis. J. Clin. Endocrinol. Metab. 2019, 104, 5737–5750. [Google Scholar] [CrossRef]
- Abreu, A.P.; Dauber, A.; Macedo, D.B.; Noel, S.D.; Brito, V.N.; Gill, J.C.; Cukier, P.; Thompson, I.R.; Navarro, V.M.; Gagliardi, P.C.; et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 2013, 368, 2467–2475. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Hayward, B.E.; Barlier, A.; Korbonits, M.; Grossman, A.B.; Jacquet, P.; Enjalbert, A.; Bonthron, D.T. Imprinting of the G(s) Alpha gene GNAS1 in the pathogenesis of acromegaly. J. Clin. Investig. 2001, 107, R31–R36. [Google Scholar] [CrossRef] [Green Version]
- Scully, K.M.; Rosenfeld, M.G. Pituitary development: Regulatory codes in mammalian organogenesis. Science 2002, 295, 2231–2235. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.Y.M.; George, A.S.; McGee, S.R.; Daly, A.Z.; Brinkmeier, M.K.; Ellsworth, B.S.; Camper, S.A. Single-cell RNA sequencing reveals novel markers of male pituitary stem cells and hormone-producing cell types. Endocrinology 2018, 159, 3910–3924. [Google Scholar] [CrossRef] [Green Version]
- Kelberman, D.; Rizzoti, K.; Lovell-Badge, R.; Robinson, I.C.A.F.; Dattani, M.T. Genetic regulation of pituitary gland development in human and mouse. Endocr. Rev. 2009, 30, 790–829. [Google Scholar] [CrossRef] [Green Version]
- Kelberman, D.; Dattani, M.T. Hypothalamic and pituitary development: Novel insights into the aetiology. Eur. J. Endocrinol. 2007, 157, S3–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzoti, K. Genetic regulation of murine pituitary development. J. Mol. Endocrinol. 2015, 54, R55–R73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, G.W. Neural control of the pituitary gland. Physiol. Rev. 1948, 28, 139–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatton, G.I. Pituicytes, glia and control of terminal secretion. J. Exp. Biol. 1988, 139, 67–79. [Google Scholar] [PubMed]
- Hatton, G.I. Emerging concepts of structure-function dynamics in adult brain: The hypothalamo-neurohypophysial system. Prog. Neurobiol. 1990, 34, 437–504. [Google Scholar] [CrossRef]
- Kopchick, J.J. History and future of growth hormone research. Horm. Res. Paediatr. 2003, 60, 103–112. [Google Scholar] [CrossRef]
- Ben-Jonathan, N.; Hnasko, R. Dopamine as a prolactin (PRL) inhibitor. Endocr. Rev. 2001, 22, 724–763. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Lazcano, I.; Sánchez-Jaramillo, E.; Uribe, R.M.; Jaimes-Hoi, L.; Joseph-Bravo, P.; Charli, J.-L. Tanycytes and the control of thyrotropin-releasing hormone flux into portal capillaries. Front. Endocrinol. 2019, 10, 401. [Google Scholar] [CrossRef] [Green Version]
- McIlwraith, E.K.; Belsham, D.D. Hypothalamic reproductive neurons communicate through signal transduction to control reproduction. Mol. Cell Endocrinol. 2020, 518, 110971. [Google Scholar] [CrossRef]
- Keller-Wood, M. Hypothalamic-pituitary—adrenal axis-feedback control. Compr. Physiol. 2015, 5, 1161–1182. [Google Scholar]
- Takuma, N.; Sheng, H.Z.; Furuta, Y.; Ward, J.M.; Sharma, K.; Hogan, B.L.; Pfaff, S.L.; Westphal, H.; Kimura, S.; Mahon, K.A. Formation of Rathke’s pouch requires dual induction from the diencephalon. Development 1998, 125, 4835–4840. [Google Scholar]
- Ericson, J.; Norlin, S.; Jessell, T.M.; Edlund, T. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development 1998, 125, 1005–1015. [Google Scholar]
- Ohuchi, H.; Horia, Y.; Yamasakia, M.; Haradab, H.; Sekinec, K.; Katoc, S.; Itoha, N. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem. Biophys. Res. Commun. 2000, 277, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Treier, M.; Gleiberman, A.S.; O’Connell, S.M.; Szeto, D.P.; McMahon, J.A.; McMahon, A.P.; Rosenfeld, M.G. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev. 1998, 12, 1691–1704. [Google Scholar] [CrossRef] [Green Version]
- Treier, M.; O’Connell, S.; Gleiberman, A.; Price, J.; Szeto, D.P.; Burgess, R.; Chuang, P.T.; McMahon, A.P.; Rosenfeld, M.G. Hedgehog signaling is required for pituitary gland development. Development 2001, 128, 377–386. [Google Scholar] [PubMed]
- Trowe, M.O.; Zhao, L.; Weiss, A.-C.; Christoffels, V.; Epstein, D.J.; Kispert, A. Inhibition of Sox2-dependent activation of Shh in the ventral diencephalon by Tbx3 is required for formation of the neurohypophysis. Development 2013, 140, 2299–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreno, G.; Apps, J.R.; Lodge, E.J.; Panousopoulos, L.; Haston, S.; Gonzalez-Meljem, J.M.; Hahn, H.; Andoniadou, C.L.; Martinez-Barbera, J.P. Hypothalamic sonic hedgehog is required for cell specification and proliferation of LHX3/LHX4 pituitary embryonic precursors. Development 2017, 144, 3289–3302. [Google Scholar] [CrossRef] [Green Version]
- Fauquier, T.; Rizzoti, K.; Dattani, M.; Lovell-Badge, R.; Robinson, I.C.A.F. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc. Natl. Acad. Sci. USA 2008, 105, 2907–2912. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Roussel-Gervais, A.; Drouin, J. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol. Cell. Biol. 2009, 29, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Japon, M.A.; Rubinstein, M.; Low, M.J. In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J. Histochem. Cytochem. 1994, 42, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamolet, B.; Pulichino, A.M.; Lamonerie, T.; Gauthier, Y.; Brue, T.; Enjalbert, A.; Drouin, J. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell 2001, 104, 849–859. [Google Scholar] [CrossRef]
- Harris, J.; Gouhier, A.; Drouin, J. Mechanisms in endocrinology: Pioneer transcription factors in pituitary development and tumorigenesis. Eur. J. Endocrinol. 2021, 184, R1–R15. [Google Scholar] [CrossRef]
- Pulichino, A.-M.; Vallette-Kasic, S.; Tsai, J.P.-Y.; Couture, C.; Gauthier, Y.; Drouin, J. Tpit determines alternate fates during pituitary cell differentiation. Genes Dev. 2003, 17, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Budry, L.; Balsalobre, A.; Gauthier, Y.; Khetchoumian, K.; L’honoré, A.; Vallette, S.; Brue, T.; Figarella-Branger, D.; Meij, B.; Drouin, J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012, 26, 2299–2310. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Crenshaw, E.B.; Rawson, E.J.; Simmons, D.M.; Swanson, L.W.; Rosenfeld, M.G. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nat. Cell Biol. 1990, 347, 528–533. [Google Scholar] [CrossRef]
- Simmons, D.M.; Voss, J.W.; Ingraham, H.A.; Holloway, J.M.; Broide, R.S.; Rosenfeld, M.G.; Swanson, L.W. Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev. 1990, 4, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Sornson, M.W.; Wu, W.; Dasen, J.S.; Flynn, S.E.; Norman, D.J.; O’Connell, S.M.; Gukovsky, I.; Carrière, C.; Ryan, A.K.; Miller, A.P.; et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nat. Cell Biol. 1996, 384, 327–333. [Google Scholar] [CrossRef]
- Gage, P.J.; Brinkmeier, M.L.; Scarlett, L.M.; Knapp, L.T.; Camper, S.A.; Mahon, K.A. The Ames dwarf gene, df, is required early in pituitary ontogeny for the extinction of Rpx transcription and initiation of lineage-specific cell proliferation. Mol. Endocrinol. 1996, 10, 1570–1581. [Google Scholar]
- Olson, L.E.; Tollkuhn, J.; Scafoglio, C.; Krones, A.; Zhang, J.; Ohgi, K.A.; Wu, W.; Taketo, M.M.; Kemler, R.; Grosschedl, R.; et al. Homeodomain-mediated β-catenin-dependent switching events dictate cell-lineage determination. Cell 2006, 125, 593–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasen, J.S.; O’Connell, S.M.; Flynn, S.E.; Treier, M.; Gleiberman, A.S.; Szeto, D.P.; Hooshmand, F.; Aggarwal, A.K.; Rosenfeld, M.G. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell 1999, 97, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Day, R.N.; Koike, S.; Sakai, M.; Muramatsu, M.; Maurer, R.A. Both Pit-1 and the estrogen receptor are required for estrogen responsiveness of the rat prolactin gene. Mol. Endocrinol. 1990, 4, 1964–1971. [Google Scholar] [CrossRef] [Green Version]
- Ellsworth, B.S.; Egashira, N.; Haller, J.L.; Butts, D.L.; Cocquet, J.; Clay, C.M.; Osamura, R.Y.; Camper, S.A. FOXL2 in the pituitary: Molecular, genetic, and developmental analysis. Mol. Endocrinol. 2006, 20, 2796–2805. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, J.; Tollkuhn, J.; Ohsawa, R.; Bresnick, E.H.; Guillemot, F.; Kageyama, R.; Rosenfeld, M.G. Sustained notch signaling in progenitors is required for sequential emergence of distinct cell lineages during organogenesis. Genes Dev. 2006, 20, 2739–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Ai, W.; Alim, Z.; Boehm, U. Embryonic gonadotropin-releasing hormone signaling is necessary for maturation of the male reproductive axis. Proc. Natl. Acad. Sci. USA 2010, 107, 16372–16377. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Bakke, M.; Krimkevich, Y.; Cushman, L.J.; Parlow, A.F.; Camper, S.A.; Parker, K.L. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development 2001, 128, 147–154. [Google Scholar]
- Rizzoti, K.; Akiyama, H.; Lovell-Badge, R. Mobilized adult pituitary stem cells contribute to endocrine regeneration in response to physiological demand. Cell Stem Cell 2013, 13, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoniadou, C.L.; Matsushima, D.; Gharavy, S.N.M.; Signore, M.; Mackintosh, A.I.; Schaeffer, M.; Gaston-Massuet, C.; Mollard, P.; Jacques, T.S.; le Tissier, P.; et al. Sox2+ stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell 2013, 13, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gremeaux, L.; Fu, Q.; Liekens, D.; van Laere, S.; Vankelecom, H. Pituitary progenitor cells tracked down by side population dissection. Stem Cells 2009, 27, 1182–1195. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Gaston-Massuet, C.; Reddy, R.; Schneider, R.P.; Blasco, M.A.; le Tissier, P.; Jacques, T.S.; Pevny, L.H.; Dattani, M.T.; Martinez-Barbera, J.P. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012, 124, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Gremeaux, L.; Luque, R.M.; Liekens, D.; Chen, J.; Buch, T.; Waisman, A.; Kineman, R.; Vankelecom, H. The adult pituitary shows stem/progenitor cell activation in response to injury and is capable of regeneration. Endocrinology 2012, 153, 3224–3235. [Google Scholar] [CrossRef]
- Russell, J.P.; Lim, X.; Santambrogio, A.; Yianni, V.; Kemkem, Y.; Wang, B.; Fish, M.; Haston, S.; Grabek, A.; Hallang, S.; et al. Pituitary stem cells produce paracrine WNT signals to control the expansion of their descendant progenitor cells. eLife 2021, 10. [Google Scholar] [CrossRef]
- Eggermann, T.; de Nanclares, G.P.; Maher, E.R.; Temple, I.K.; Tümer, Z.; Monk, D.; Mackay, D.J.G.; Grønskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting disorders: A group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 2015, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Delgado, M.; Riccio, A.; Eggermann, T.; Maher, E.R.; Lapunzina, P.; Mackay, D.; Monk, D. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016, 32, 444–455. [Google Scholar] [CrossRef] [Green Version]
- Mussa, A.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: A paradigm for genomic medicine. Clin. Genet. 2016, 89, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Vilain, E.; le Merrer, M.; Lecointre, C.; Desangles, F.; Kay, M.A.; Maroteaux, P.; McCabe, E.R.B. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J. Clin. Endocrinol. Metab. 1999, 84, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.B.; Day, F.; Elks, C.E.; Sulem, P.; Thompson, D.J.; Ferreira, T.; He, C.; Chasman, D.I.; Esko, T.; Thorleifsson, G.; et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nat. Cell Biol. 2014, 514, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Davies, W.; Lynn, P.M.; Relkovic, D.; Wilkinson, L.S. Imprinted genes and neuroendocrine function. Front. Neuroendocr. 2008, 29, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Arima, T.; Kamikihara, T.; Hayashida, T.; Kato, K.; Inoue, T.; Shirayoshi, Y.; Oshimura, M.; Soejima, H.; Mukai, T.; Wake, N. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005, 33, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef]
- Adhami, H.A.; Evano, B.; le Digarcher, A.; Gueydan, C.; Dubois, E.; Parrinello, H.; Dantec, C.; Bouschet, T.; Varrault, A.; Journot, L. A systems-level approach to parental genomic imprinting: The imprinted gene network includes extracellular matrix genes and regulates cell cycle exit and differentiation. Genome Res. 2015, 25, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varrault, A.; Dantec, C.; le Digarcher, A.; Chotard, L.; Bilanges, B.; Parrinello, H.; Dubois, E.; Rialle, S.; Severac, D.; Bouschet, T.; et al. Identification of Plagl1/Zac1 binding sites and target genes establishes its role in the regulation of extracellular matrix genes and the imprinted gene network. Nucleic Acids Res. 2017, 45, 10466–10480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabory, A.; Ripoche, M.-A.; le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forne, T.; Jammes, H.; Ainscough, J.F.X.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef]
- Lui, J.C.; Finkielstain, G.P.; Barnes, K.M.; Baron, J. An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R189–R196. [Google Scholar] [CrossRef]
- Boucher, J.; Charalambous, M.; Zarse, K.; Mori, M.A.; Kleinridders, A.; Ristow, M.; Ferguson-Smith, A.C.; Kahn, C.R. Insulin and insulin-like growth factor 1 receptors are required for normal expression of imprinted genes. Proc. Natl. Acad. Sci. USA 2014, 111, 14512–14517. [Google Scholar] [CrossRef] [Green Version]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Ferrón, S.R.; Radford, E.J.; Domingo-Muelas, A.; Kleine, I.; Ramme, A.; Gray, D.; Sandovici, I.; Constancia, M.; Ward, A.; Menheniott, T.R.; et al. Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat. Commun. 2015, 6, 8265. [Google Scholar] [CrossRef] [Green Version]
- Ferrón, S.R.; Charalambous, M.; Radford, E.; McEwen, K.; Wildner, H.; Hind, E.; Morante-Redolat, J.M.; Laborda, J.; Guillemot, F.; Bauer, S.R.; et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nat. Cell Biol. 2011, 475, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Martinet, C.; Monnier, P.; Louault, Y.; Benard, M.; Gabory, A.; Dandolo, L. H19controls reactivation of the imprinted gene network during muscle regeneration. Development 2016, 143, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Correra, R.M.; Ollitrault, D.; Valente, M.; Mazzola, A.; Adalsteinsson, B.T.; Ferguson-Smith, A.C.; Marazzi, G.; Sassoon, D.A. The imprinted gene Pw1/Peg3 regulates skeletal muscle growth, satellite cell metabolic state, and self-renewal. Sci. Rep. 2018, 8, 14649. [Google Scholar] [CrossRef] [PubMed]
- Besson, V.; Smeriglio, P.; Wegener, A.; Relaix, F.; Oumesmar, B.N.; Sassoon, D.A.; Marazzi, G. PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proc. Natl. Acad. Sci. USA 2011, 108, 11470–11475. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.S.; Lin, K.K.; Sonnet, C.; Boles, N.C.; Weksberg, D.C.; Nguyen, H.; Holt, L.J.; Rickwood, D.; Daly, R.J.; Goodell, M.A. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS ONE 2011, 6, e26410. [Google Scholar] [CrossRef]
- Grattan, D.R.; Steyn, F.J.; Kokay, I.C.; Anderson, G.M.; Bunn, S.J. Pregnancy-induced adaptation in the neuroendocrine control of prolactin secretion. J. Neuroendocr. 2008, 20, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.E.; Kanyicska, B.; Lerant, A.; Nagy, G. Prolactin: Structure, function, and regulation of secretion. Physiol. Rev. 2000, 80, 1523–1631. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Cano, M.; Landefeld, T.D. The effects of estrogens on tumor growth and on prolactin and growth hormone mRNA expression in rat pituitary tissues. Am. J. Pathol. 1988, 133, 397–406. [Google Scholar]
- Asa, S.L.; Penz, G.; Kovacs, K.; Ezrin, C. Prolactin cells in the human pituitary. A quantitative immunocytochemical analysis. Arch. Pathol. Lab. Med. 1982, 106, 360–363. [Google Scholar]
- Castrique, E.; Fernandez-Fuente, M.; le Tissier, P.; Herman, A.; Levy, A. Use of a prolactin-Cre/ROSA-YFP transgenic mouse provides no evidence for lactotroph transdifferentiation after weaning, or increase in lactotroph/somatotroph proportion in lactation. J. Endocrinol. 2010, 205, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kasti, M.M.; Christian, H.C.; Huerta-Ocampo, I.; Stolbrink, M.; Gill, S.; Houston, P.A.; Davies, J.; Chilcott, J.; Hill, N.; Matthews, D.R.; et al. The pregnancy-induced increase in baseline circulating growth hormone in rats is not induced by Ghrelin. J. Neuroendocr. 2008, 20, 309–322. [Google Scholar] [CrossRef]
- Cleaton, M.A.M.; Dent, C.L.; Howard, M.; Corish, J.A.; Gutteridge, I.; Sovio, U.M.; Gaccioli, F.; Takahashi, N.; Bauer, S.R.; Charnock-Jones, D.S.; et al. Fetus-derived DLK1 is required for maternal metabolic adaptations to pregnancy and is associated with fetal growth restriction. Nat. Genet. 2016, 48, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, F.C.; Charalambous, M. Genomic imprinting, growth and maternal-fetal interactions. J. Exp. Biol. 2018, 221, jeb164517. [Google Scholar] [CrossRef] [Green Version]
- Creeth, H.; McNamara, G.; Isles, A.; John, R. Imprinted genes influencing the quality of maternal care. Front. Neuroendocr. 2019, 53, 100732. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.; Garfield, A.S.; Madon-Simon, M.; Charalambous, M.; Clarkson, R.W.; Smalley, M.J.; Kendrick, H.; Isles, A.R.; Parry, A.J.; Carney, S.; et al. Developmental programming mediated by complementary roles of imprinted Grb10 in mother and pup. PLoS Biol. 2014, 12, e1001799. [Google Scholar] [CrossRef] [Green Version]
- Giles, A.; Madec, F.; Friedrichsen, S.; Featherstone, K.; Chambers, T.; Harper, C.V.; Resch, J.; Brabant, G.; Davis, J.R.E. Wnt signaling in estrogen-induced lactotroph proliferation. J. Cell Sci. 2011, 124, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Babak, T.; DeVeale, B.; Tsang, E.K.; Zhou, Y.; Li, X.; Smith, K.S.; Kukurba, K.R.; Zhang, R.; Li, J.B.; Van Der Kooy, D.; et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 2015, 47, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Ellis, P.; Fagan, B.M.; Magness, S.T.; Hutton, S.; Taranova, O.; Hayashi, S.; McMahon, A.; Rao, M.; Pevny, L. SOX2, a Persistent Marker for Multipotential Neural Stem Cells Derived from Embryonic Stem Cells, the Embryo or the Adult. Dev. Neurosci. 2004, 26, 148–165. [Google Scholar] [CrossRef]
- Lamble, S.; Batty, E.; Attar, M.; Buck, D.; Bowden, R.; Lunter, G.; Crook, D.; El-Fahmawi, B.; Piazza, P. Improved workflows for high throughput library preparation using the transposome-based nextera system. BMC Biotechnol. 2013, 13, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, J. Eulerr: Area-Proportional Euler and Venn diagrams With Ellipses, R package version 6.1.0; Lund University: Lund, Sweden, 2020. [Google Scholar]
- Carlson, M. LumiMouseAll.db, R package version 1.22.0. Illumina Mouse Illumina expression annotation data (chip lumiMouseAll); Illumina: San Diego, CA, USA, 2013. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Giri, D.; Vignola, M.L.; Gualtieri, A.; Scagliotti, V.; McNamara, P.; Peak, M.; Didi, M.; Gaston-Massuet, C.; Senniappan, S. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum. Mol. Genet. 2017, 26, 4315–4326. [Google Scholar] [CrossRef]
- Charalambous, M.; Cowley, M.; Geoghegan, F.; Smith, F.M.; Radford, E.J.; Marlow, B.P.; Graham, C.F.; Hurst, L.D.; Ward, A. Maternally-inherited Grb10 reduces placental size and efficiency. Dev. Biol. 2010, 337, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, M.; da Rocha, S.T.; Ferguson-Smith, A.C. Genomic imprinting, growth control and the allocation of nutritional resources: Consequences for postnatal life. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.B.; Bartolomei, M.S. Regulation of imprinting in clusters: Noncoding RNAs versus insulators. Adv. Genet. 2008, 61, 207–223. [Google Scholar] [PubMed]
- Cheung, L.Y.M.; Camper, S.A. PROP1-dependent retinoic acid signaling regulates developmental pituitary morphogenesis and hormone expression. Endocrinology 2020, 161. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; McCole, R.B.; Woodfine, K.; Wood, A.J.; Chahal, M.; Monk, D.; Moore, G.E.; Oakey, R.J. Transcript- and tissue-specific imprinting of a tumour suppressor gene. Hum. Mol. Genet. 2008, 18, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, L.L.; Haisenleder, D.J.; Aylor, K.W.; Marshall, J.C. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol. Reprod. 2009, 81, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, M.T.; Boldt, H.B.; Laursen, L.S.; Sottrup-Jensen, L.; Conover, C.A.; Oxvig, C. Pregnancy-associated plasma protein-A2 (PAPP-A2), a novel insulin-like growth factor-binding Protein-5 proteinase. J. Biol. Chem. 2001, 276, 21849–21853. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; He, H.; Liu, Q.; Zhang, F.; Lv, J.; Zeng, T.; Gu, N.; Wu, Q. Identification and epigenetic analysis of a maternally imprinted gene Qpct. Mol. Cells 2015, 38, 859–865. [Google Scholar]
- Menheniott, T.R.; Woodfine, K.; Schulz, R.; Wood, A.J.; Monk, D.; Giraud, A.S.; Baldwin, H.S.; Moore, G.E.; Oakey, R.J. Genomic imprinting of Dopa decarboxylase in heart and reciprocal allelic expression with neighboring Grb10. Mol. Cell Biol. 2008, 28, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Kuroiwa, Y.; Kohda, T.; Shitara, H.; Yonekawa, H.; Kawabe, T.; Hasegawa, H.; Barton, S.C.; Surani, M.A.; Kaneko-Ishino, T.; et al. Identification of the Meg1/Grb10 imprinted gene on mouse proximal chromosome 11, a candidate for the Silver–Russell syndrome gene. Proc. Natl. Acad. Sci. USA 1998, 95, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, P.; Monk, D.; Hitchins, M.; Gordon, E.; Dean, W.; Beechey, C.V.; Peters, J.; Craigen, W.; Preece, M.; Stanier, P.; et al. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum. Mol. Genet. 2003, 12, 1005–1019. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Roth, R.A. Grb-IR: A SH2-domain-containing protein that binds to the insulin receptor and inhibits its function. Proc. Natl. Acad. Sci. USA 1995, 92, 10287–10291. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yoon, S.-O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Burks, D.J.; De Mora, J.F.; Schubert, M.; Withers, D.J.; Myers, M.G.; Towery, H.H.; Altamuro, S.L.; Flint, C.L.; White, M.F. IRS-2 pathways integrate female reproduction and energy homeostasis. Nat. Cell Biol. 2000, 407, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Williams, J.; Sabers, C.; Wiederrecht, G.; Lawrence, J.C.; Abraham, R.T. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996, 15, 5256–5267. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sáiz, V.; Targosz, B.-S.; Lemeer, S.; Eichner, R.; Langer, C.; Bullinger, L.; Reiter, C.; Slotta-Huspenina, J.; Schroeder, S.; Knorn, A.-M.; et al. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nat. Cell Biol. 2013, 15, 72–81. [Google Scholar] [CrossRef]
- Christenson, J.G.; Dairman, W.; Udenfriend, S. On the identity of DOPA decarboxylase and 5-hydroxytryptophan decarbox-ylase (immunological titration-aromatic L-amino acid decarboxylase-serotonin-dopamine-norepinephrine). Proc. Natl. Acad. Sci. USA 1972, 69, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Ida, T.; Fudetani, M.; Mori, M.; Kaiya, H.; Hino, J.; Nakahara, K.; Murakami, N.; Miyazato, M.; Kangawa, K. Identification of neuromedin U precursor-related peptide and its possible role in the regulation of prolactin release. Sci. Rep. 2017, 7, 10468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacicedo, L.; Ortego, J.; Melian, E.; Sánchez-Franco, F. Prolactin gene expression and secretion during pregnancy and lactation in the rat: Role of dopamine and vasoactive intestinal peptide. Endocrinology 1996, 137, 631–637. [Google Scholar]
- DeChiara, T.M.; Efstratiadis, A.; Robertsen, E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nat. Cell Biol. 1990, 345, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, M.; Smith, F.M.; Bennett, W.R.; Crew, T.E.; MacKenzie, F.; Ward, A. Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc. Natl. Acad. Sci. USA 2003, 100, 8292–8297. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.M.; Holt, L.J.; Garfield, A.S.; Charalambous, M.; Koumanov, F.; Perry, M.; Bazzani, R.; Sheardown, S.A.; Hegarty, B.D.; Lyons, R.J.; et al. Mice with a Disruption of the Imprinted Grb10 Gene Exhibit Altered Body Composition, Glucose Homeostasis, and Insulin Signaling during Postnatal Life. Mol. Cell. Biol. 2007, 27, 5871–5886. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Reynisdottir, I.; Massague, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995, 9, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Edwards, M.C.; Bai, C.; Parker, S.; Zhang, P.; Baldini, A.; Harper, J.W.; Elledge, S.J. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995, 9, 650–662. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liégeois, N.J.; Wong, C.; Finegold, M.J.; Hou, H.; Thompson, J.C.; Silverman, A.; Harper, J.W.; Depinho, R.A.; Elledge, S.J. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith–Wiedemann syndrome. Nat. Cell Biol. 1997, 387, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.C.; Wood, M.D.; Tunster, S.J.; Barton, S.C.; Surani, A.M.; John, R.M. Cdkn1c (p57Kip2) is the major regulator of embryonic growth within its imprinted domain on mouse distal chromosome 7. BMC Dev. Biol. 2007, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curley, J.P.; Barton, S.; Surani, A.; Keverne, E.B. Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc. R. Soc. B Boil. Sci. 2004, 271, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Denizot, A.-L.; Besson, V.; Correra, R.M.; Mazzola, A.; Lopes, I.; Courbard, J.-R.; Marazzi, G.; Sassoon, D.A. A Novel Mutant Allele of Pw1/Peg3 Does Not Affect Maternal Behavior or Nursing Behavior. PLoS Genet. 2016, 12, e1006053. [Google Scholar] [CrossRef] [PubMed]
- Li, L. Regulation of Maternal Behavior and Offspring Growth by Paternally Expressed Peg3. Science 1999, 284, 330–334. [Google Scholar] [CrossRef]
- Valente, T.; Junyent, F.; Auladell, C. Zac1 is expressed in progenitor/stem cells of the neuroectoderm and mesoderm during embryogenesis: Differential phenotype of the Zac1-expressing cells during development. Dev. Dyn. 2005, 233, 667–679. [Google Scholar] [CrossRef]
- Pagotto, U.; Arzberger, T.; Ciani, E.; Lezoualc’h, F.; Pilon, C.; Journot, L.; Spengler, D.; Stalla, G.K. Inhibition of Zac1, a new gene differentially expressed in the anterior pituitary, increases cell proliferation. Endocrinology 1999, 140, 987–996. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Tichomirowa, M.A.; Sievers, C.; Yassouridis, A.; Arzberger, T.; Hougrand, O.; Deprez, M.; Daly, A.F.; Petrossians, P.; Pagotto, U.; et al. Tumor ZAC1 expression is associated with the response to somatostatin analog therapy in patients with acromegaly. Int. J. Cancer 2009, 125, 2122–2126. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Glaser, J.; Borsos, M.; El Marjou, F.; Walter, M.; Teissandier, A.; Bourc’His, D. Transient transcription in the early embryo sets an epigenetic state that programs postnatal growth. Nat. Genet. 2016, 49, 110–118. [Google Scholar] [CrossRef]
- Braxton, T.M.; Sarpong, D.E.A.; Dovey, J.L.; Guillou, A.; Evans, B.A.J.; Castellano, J.M.; Keenan, B.E.; Baraghithy, S.; Evans, S.L.; Tena-Sempere, M.; et al. Thermoneutrality improves skeletal impairment in adult Prader–Willi syndrome mice. J. Endocrinol. 2019, 243, 175–186. [Google Scholar] [CrossRef]
- Costa, R.A.; Ferreira, I.R.; Cintra, H.A.; Gomes, L.H.F.; Guida, L.D.C. Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome. Front. Endocrinol. 2019, 10, 864. [Google Scholar] [CrossRef] [Green Version]
- Resnick, J.L.; Nicholls, R.D.; Wevrick, R. Recommendations for the investigation of animal models of Prader–Willi syndrome. Mamm. Genome 2013, 24, 165–178. [Google Scholar] [CrossRef]
- Tennese, A.A.; Wevrick, R. Impaired Hypothalamic Regulation of Endocrine Function and Delayed Counterregulatory Response to Hypoglycemia in Magel2-Null Mice. Endocrinology 2011, 152, 967–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; HogenEsch, J.B.; Muglia, L.J.; Van Gelder, R.N.; Herzog, E.D.; Stewart, C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.E.; Wevrick, R. Loss of Magel2, a Candidate Gene for Features of Prader-Willi Syndrome, Impairs Reproductive Function in Mice. PLOS ONE 2009, 4, e4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, N.L.; Wevrick, R.; Mellon, P.L. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 2008, 18, 248–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, T.; Zimmer, M.; Mugele, K.; Spiess, J. Primary structure and functional expression of a glutaminyl cyclase. Proc. Natl. Acad. Sci. USA 1991, 88, 10059–10063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, S.; Kohlmann, S.; Bäuscher, C.; Sedlmeier, R.; Koch, B.; Eichentopf, R.; Becker, A.; Cynis, H.; Hoffmann, T.; Berg, S.; et al. Glutaminyl cyclase knock-out mice exhibit slight hypothyroidism but no hypogonadism: Implications for enzyme function and drug development. J. Biol. Chem. 2011, 286, 14199–14208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiura, H.; Nakamura, K.; Hikichi, T.; Hino, T.; Oda, K.; Suzuki-Migishima, R.; Kohda, T.; Kaneko-ishino, T.; Ishino, F. Paternal deletion of Meg1/Grb10 DMR causes maternalization of the Meg1/Grb10 cluster in mouse proximal Chromosome 11 leading to severe pre- and postnatal growth retardation. Hum. Mol. Genet. 2009, 18, 1424–1438. [Google Scholar] [CrossRef] [Green Version]
- Monk, D.; Arnaud, P.; Frost, J.; Hills, F.A.; Stanier, P.; Feil, R.; Moore, G.E. Reciprocal imprinting of human GRB10 in placental trophoblast and brain: Evolutionary conservation of reversed allelic expression. Hum. Mol. Genet. 2009, 18, 3066–3074. [Google Scholar] [CrossRef]
- Orrillo, S.J.; de Dio, N.; Asad, A.S.; De Fino, F.; Imsen, M.; Romero, A.C.; Zárate, S.; Ferrari, J.; Pisera, D. Anterior pituitary gland synthesises dopamine from l-3,4-dihydroxyphenylalanine (l-dopa). J. Neuroendocrinol. 2020, 32, e12885. [Google Scholar] [CrossRef]
- Baran, Y.; Subramaniam, M.; Biton, A.; Tukiainen, T.; Tsang, E.K.; Rivas, M.A.; Pirinen, M.; Gutierrez-Arcelus, M.; Smith, K.S.; Kukurba, K.R.; et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015, 25, 927–936. [Google Scholar] [CrossRef] [Green Version]




| Location of Imprinting Cluster | Type of Mutation | Genes Affected | Normally Expressed Allele | Syndrome | Main Endocrine Features | Other Main Features | Ref | ||
|---|---|---|---|---|---|---|---|---|---|
| Growth | Sexual Development | Metabolic Conditions | |||||||
| 6q24 | UPD(6)pat/duplication of paternal allele/Hypomethylation of maternal DMR | PLAGL1 | Paternal | Transient Neonatal Diabetes Mellitus [OMIM 601410] | Severe IUGR | Not reported | Hyperglycaemia, dehydration, absence of ketoacidosis | [6,7,8,9,10] | |
| HYMAI | |||||||||
| 7p11.2-q13 | UPD(7)mat | GRB10 | Maternal | Silver–Russell [OMIM 180860] | IUGR, relative macrocephaly, postnatal growth failure | Premature adrenarche. Early and rapid puberty. | Perinatal feeding difficulties and hypoglycaemia. Develop insulin resistance. | Distinctive facial features (triangular shape, prominent forehead, narrow chin, small jaw). Clinodactyly. | [11,12,13,14,15,16,17] |
| 11p15.5 | Hypomethylation of IC1 | IGF-2 | Paternal | ||||||
| H19 | Maternal | ||||||||
| Loss-of-function mutations | IGF-2 | Paternal | |||||||
| Gain of methylation IC2 | CDKN1C | Maternal | |||||||
| KCNQ1 | Maternal | ||||||||
| KCNQ1OT1 | Paternal | ||||||||
| 11p15.5 | Gain of methylation IC1 | IGF-2 | Paternal | Beckwith–Wiedemann [OMIM 130650] | Pre- and postnatal overgrowth. | Not reported | Neonatal hyperinsulinism | Macroglossia, abdominal wall defects. Predisposition to tumour development (Wilm’s tumour, adrenal carcinoma, hepatoblastoma) early in life. Visceromegaly. Renal abnormalities. | [18,19,20,21,22,23,24,25] |
| H19 | Maternal | ||||||||
| Loss of methylation IC2 | CDKN1C | Maternal | |||||||
| KCNQ1 | Maternal | ||||||||
| KCNQ1OT1 | Paternal | ||||||||
| Loss-of-function mutations | CDKN1C | Maternal | |||||||
| 11p15.5 | Gain-of-function missense mutations | CDKN1C | Maternal | IMAGe [OMIM 614732] | IUGR | Genital abnormalities in males (micropenis, cryptorchidism, hypospadias). | Metaphyseal dysplasia. Adrenal insufficiency. Skeletal abnormalities | [26] | |
| 11p15.5 | Missense mutations | KCNQ1 | Maternal | Growth hormone deficiency (GHD) and gingival fibromatosis [OMIM 611010] | Small stature | Gonadotrophin deficiency | Gingival fibromatosis. | [27] | |
| 14q32 | UPD14)mat | DLK1 RTL1 DIO3 MEG3 MEG8 | Paternal Maternal | Temple [OMIM 616222] | IUGR. Postnatal short stature | Premature sexual development | Feeding difficulties in the neonatal period. Truncal obesity | Muscular hypotonia, motor and mental developmental delay, scoliosis | [28] |
| UPD14)pat | Kagami–Ogata [OMIM 608149] | Postnatal growth retardation | [29] | ||||||
| 14q32 | Inactivating mutations, deletions | DLK1 | Paternal | Central precocious puberty | Premature sexual development | Truncal overweight/obesity, insulin resistance, T2DM, hyperlipidaemia | [30] | ||
| 15q11-q13 | Deletion of paternal region/mUPD | MKRN3 MAGEL2 NDN NPAP1 SNRPN SNORD116 | Paternal | Prader–Willi [OMIM 176270] | Short stature. Reduced GH. Reduced IGF-I Low bone density. | Variable hypogonadism phenotype (genital hypoplasia, incomplete pubertal development, infertility) Low to normal levels of testosterone or oestrogen, FSH and LH. GnRH insensitivity | Hyperphagia | Mild intellectual disability, obsessive-compulsive traits | [31,32,33,34,35] |
| 15q11-q13 | Deletion of maternal region/UPD(15)pat | UBE3A | Maternal | Angelman [OMIM 105830] | Intellectual disability, Speech impairment, gait ataxia | [36,37] | |||
| 15q11-q13 | Point mutations, deletions, frameshifts | MAGEL2 | Schaaf–Yang [OMIM 615547] | GHD. Short stature. | Gonadotrophin deficiency | Hyperinsulinaemic hypoglycaemia. Neonatal feeding difficulties, followed by hyperphagia. Central hypothyroidism | Panhypopituitarism associated with a hypoplastic anterior pituitary gland. Adrenal insufficiency. Arthrogryposis. ASD. Intellectual disability | [38,39] | |
| 15q11-q13 | Inactivating mutations | MKRN3 | Paternal | Central precocious puberty | Premature reactivation of the reproductive axis. | [40] | |||
| 20q13.2-13.3 | Activating mutations | GNAS | McCune–Albright [OMIM 174800] | Acromegaly (caused by GH-secreting pituitary tumours) | Sexual precocity | Hyperthyroidism (caused by hyperactive thyroid nodules) Hypercortisolism (associated with macronodular adrenal hyperplasia or adrenal adenomas) | Hyperpigmentation of the skin Osteomalacia | [41,42] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scagliotti, V.; Esse, R.; Willis, T.L.; Howard, M.; Carrus, I.; Lodge, E.; Andoniadou, C.L.; Charalambous, M. Dynamic Expression of Imprinted Genes in the Developing and Postnatal Pituitary Gland. Genes 2021, 12, 509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040509
Scagliotti V, Esse R, Willis TL, Howard M, Carrus I, Lodge E, Andoniadou CL, Charalambous M. Dynamic Expression of Imprinted Genes in the Developing and Postnatal Pituitary Gland. Genes. 2021; 12(4):509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040509
Chicago/Turabian StyleScagliotti, Valeria, Ruben Esse, Thea L. Willis, Mark Howard, Isabella Carrus, Emily Lodge, Cynthia L. Andoniadou, and Marika Charalambous. 2021. "Dynamic Expression of Imprinted Genes in the Developing and Postnatal Pituitary Gland" Genes 12, no. 4: 509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040509