
Machine Learning Modeling from Omics Data as Prospective Tool for Improvement of Inflammatory Bowel Disease Diagnosis and Clinical Classifications

 , , and
, , and
Abstract
:1. Introduction
2. Machine Learning Approaches
2.1. Fitting the Model
2.2. Linear Algorithms
2.3. Nonlinear Algorithms
2.4. Clustering Algorithms
3. Machine Learning in IBD Research
3.1. Machine Learning Using Genomic Data
3.2. Machine Learning Using Transcriptomic Data
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef]
- Ruel, J.; Ruane, D.; Mehandru, S.; Gower-Rousseau, C.; Colombel, J.F. IBD across the age spectrum—Is it the same disease? Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 88–98. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Shi, H.Y.; Tang, W.; Law, C.C.Y.; Sung, J.J.Y.; Chan, F.K.L.; Ng, S.C. Systematic review and meta-analysis: Phenotype and clinical outcomes of older-onset inflammatory bowel disease. J. Crohn’s Colitis 2016, 10, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.R.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.F.; Gasche, C.; Geboes, K.; et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19 (Suppl. A), 5A–36A. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative Colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [Green Version]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Levine, A.; Koletzko, S.; Turner, D.; Escher, J.C.; Cucchiara, S.; De Ridder, L.; Kolho, K.L.; Veres, G.; Russell, R.K.; Paerregaard, A.; et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Seyed Tabib, N.S.; Madgwick, M.; Sudhakar, P.; Verstockt, B.; Korcsmaros, T.; Vermeire, S. Big data in IBD: Big progress for clinical practice. Gut 2020, 69, 1520–1532. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.B.; Kugathasan, S.; Cho, J.H. Genetics of Inflammatory Bowel Diseases. Gastroenterology 2015, 149, 1163–1176.e2. [Google Scholar] [CrossRef] [Green Version]
- Glocker, E.-O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Z.; Van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Stappenbeck, T.S.; Rioux, J.D.; Mizoguchi, A.; Saitoh, T.; Huett, A.; Darfeuille-Michaud, A.; Wileman, T.; Mizushima, N.; Carding, S.; Akira, S.; et al. Crohn disease: A current perspective on genetics, autophagy and immunity. Autophagy 2011, 7, 355–374. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Vande Casteele, N. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Furey, T.S.; Sethupathy, P.; Sheikh, S.Z. Redefining the IBDs using genome-scale molecular phenotyping. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 296–311. [Google Scholar] [CrossRef]
- Mishra, R.; Dhawan, P.; Srivastava, A.S.; Singh, A.B. Inflammatory bowel disease: Therapeutic limitations and prospective of the stem cell therapy. World J. Stem Cells 2020, 12, 1050–1066. [Google Scholar] [CrossRef]
- Henderson, P.; Van Limbergen, J.E.; Schwarze, J.; Wilson, D.C. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 382–395. [Google Scholar] [CrossRef]
- Dyson, J.K.; Rutter, M.D. Colorectal cancer in inflammatory bowel disease: What is the real magnitude of the risk? World J. Gastroenterol. 2012, 18, 3839–3848. [Google Scholar] [CrossRef]
- Tran, V.; Shammas, R.M.; Sauk, J.S.; Padua, D. Evaluating tofacitinib citrate in the treatment of moderate-to-severe active ulcerative colitis: Design, development and positioning of therapy. Clin. Exp. Gastroenterol. 2019, 12, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogler, G. Gastrointestinal and liver adverse effects of drugs used for treating IBD. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Adegbola, S.O.; Sahnan, K.; Warusavitarne, J.; Hart, A.; Tozer, P. Anti-TNF therapy in Crohn’s disease. Int. J. Mol. Sci. 2018, 19, 2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, A.; Holzwanger, E.A.; Levy, A.N. Emerging use of artificial intelligence in inflammatory bowel disease. World J. Gastroenterol. 2020, 26, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Brookes, M.J.; Green, J.R.B. Maintenance of remission in Crohn’s disease: Current and emerging therapeutic options. Drugs 2004, 64, 1069–1089. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, S.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Meltzer, S.J.; Brant, S.R.; Kwon, J.H. Identification of microRNAs associated with ileal and colonic Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Dulai, P.S.; Singh, S.; Vande Casteele, N.; Boland, B.S.; Rivera-Nieves, J.; Ernst, P.B.; Eckmann, L.; Barrett, K.E.; Chang, J.T.; Sandborn, W.J. Should We Divide Crohn’s Disease Into Ileum-Dominant and Isolated Colonic Diseases? Clin. Gastroenterol. Hepatol. 2019, 17, 2634–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häsler, R.; Feng, Z.; Bäckdahl, L.; Spehlmann, M.E.; Franke, A.; Teschendorff, A.; Rakyan, V.K.; Down, T.A.; Wilson, G.A.; Feber, A.; et al. A functional methylome map of ulcerative colitis. Genome Res. 2012, 22, 2130–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.C.E.; Weiser, M.; Lee, S.E.; Gipson, G.R.; Iyer, V.B.; Sartor, R.B.; Herfarth, H.H.; Long, M.D.; Hansen, J.J.; Isaacs, K.L.; et al. MicroRNAs classify different disease behavior phenotypes of Crohn’s disease and may have prognostic utility. Inflamm. Bowel Dis. 2015, 21, 2178–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigorta, U.M.; Denson, L.A.; Hyams, J.S.; Mondal, K.; Prince, J.; Walters, T.D.; Griffiths, A.; Noe, J.D.; Crandall, W.V.; Rosh, J.R.; et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 2017, 49, 1517–1521. [Google Scholar] [CrossRef]
- Romagnoni, A.; Jégou, S.; Van Steen, K.; Wainrib, G.; Hugot, J.P.; Peyrin-Biroulet, L.; Chamaillard, M.; Colombel, J.F.; Cottone, M.; D’Amato, M.; et al. Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data. Sci. Rep. 2019, 9, 10351. [Google Scholar] [CrossRef]
- Gubatan, J.; Levitte, S.; Patel, A.; Balabanis, T.; Wei, M.T.; Sinha, S.R. Artificial intelligence applications in inflammatory bowel disease: Emerging technologies and future directions. World J. Gastroenterol. 2021, 27, 1920–1935. [Google Scholar] [CrossRef]
- Stankovic, B.; Dragasevic, S.; Popovic, D.; Zukic, B.; Kotur, N.; Sokic-Milutinovic, A.; Alempijevic, T.; Lukic, S.; Milosavljevic, T.; Nikcevic, G.; et al. Variations in inflammatory genes as molecular markers for prediction of inflammatory bowel disease occurrence. J. Dig. Dis. 2015, 16, 723–733. [Google Scholar] [CrossRef]
- Waljee, A.K.; Lipson, R.; Wiitala, W.L.; Zhang, Y.; Liu, B.; Zhu, J.; Wallace, B.; Govani, S.M.; Stidham, R.W.; Hayward, R.; et al. Predicting Hospitalization and Outpatient Corticosteroid Use in Inflammatory Bowel Disease Patients Using Machine Learning. Inflamm. Bowel Dis. 2017, 24, 45–53. [Google Scholar] [CrossRef]
- Waljee, A.K.; Joyce, J.C.; Wang, S.; Saxena, A.; Hart, M.; Zhu, J.; Higgins, P.D.R. Algorithms outperform metabolite tests in predicting response of patients with inflammatory bowel disease to thiopurines. Clin. Gastroenterol. Hepatol. 2010, 8, 143–150. [Google Scholar] [CrossRef]
- Waljee, A.K.; Liu, B.; Sauder, K.; Zhu, J.; Govani, S.M.; Stidham, R.W.; Higgins, P.D.R. Predicting corticosteroid-free endoscopic remission with vedolizumab in ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Waljee, A.K.; Liu, B.; Sauder, K.; Zhu, J.; Govani, S.M.; Stidham, R.W.; Higgins, P.D.R. Predicting Corticosteroid-Free Biologic Remission with Vedolizumab in Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 1185–1192. [Google Scholar] [CrossRef]
- Mossotto, E.; Ashton, J.J.; Coelho, T.; Beattie, R.M.; MacArthur, B.D.; Ennis, S. Classification of Paediatric Inflammatory Bowel Disease using Machine Learning. Sci. Rep. 2017, 7, 2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, N.; Sohrabi, K.; Schneider, H.; Zimmer, K.-P.; Fischer, P.; de Laffolie, J. Machine Learning Classification of Inflammatory Bowel Disease in Children Based on a Large Real-World Pediatric Cohort CEDATA-GPGE® Registry. Front. Med. 2021, 8, 666190. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Ishihara, S.; Fujishiro, M.; Saito, H.; Kumagai, Y.; Shichijo, S.; Aoyama, K.; Tada, T. Novel computer-assisted diagnosis system for endoscopic disease activity in patients with ulcerative colitis. Gastrointest. Endosc. 2019, 89, 416–421.e1. [Google Scholar] [CrossRef]
- Stidham, R.W.; Liu, W.; Bishu, S.; Rice, M.D.; Higgins, P.D.R.; Zhu, J.; Nallamothu, B.K.; Waljee, A.K. Performance of a Deep Learning Model vs Human Reviewers in Grading Endoscopic Disease Severity of Patients With Ulcerative Colitis. JAMA Netw. Open 2019, 2, e193963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, J. Master Machine Learning Algorithms: Discover how they work and implement them from scratch. In Machine Learning Mastery, 5th ed.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Isakov, O.; Dotan, I.; Ben-Shachar, S. Machine Learning-Based Gene Prioritization Identifies Novel Candidate Risk Genes for Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lai, L.; Shen, J. Development of a susceptibility gene based novel predictive model for the diagnosis of ulcerative colitis using random forest and artificial neural network. Aging (Albany. NY) 2020, 12, 20471–20482. [Google Scholar] [CrossRef]
- Pal, L.R.; Kundu, K.; Yin, Y.; Moult, J. CAGI4 Crohn’s exome challenge: Marker SNP versus exome variant models for assigning risk of Crohn disease. Hum. Mutat. 2017, 38, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Cushing, K.C.; McLean, R.; McDonald, K.G.; Gustafsson, J.K.; Knoop, K.A.; Kulkarni, D.H.; Sartor, R.B.; Newberry, R.D. Predicting risk of postoperative disease recurrence in Crohn’s disease: Patients with indolent Crohn’s disease have distinct whole transcriptome profiles at the time of first surgery. Inflamm. Bowel Dis. 2019, 25, 180–193. [Google Scholar] [CrossRef]
- Khorasani, H.M.; Usefi, H.; Peña-Castillo, L. Detecting ulcerative colitis from colon samples using efficient feature selection and machine learning. Sci. Rep. 2020, 10, 13744. [Google Scholar] [CrossRef]
- Shai, S.-S.; Shai, B.-D. UNDERSTANDING MACHINE LEARNING—From Theory to Algorithms; Cambridge University Press: New York, NY, USA, 2014; pp. 114–123. [Google Scholar]
- Melo, F. Area under the ROC Curve. In Encyclopedia of Systems Biology; Springer New York: New York, NY, USA, 2013; pp. 38–39. [Google Scholar]
- Smith, A.M.; Walsh, J.R.; Long, J.; Davis, C.B.; Henstock, P.; Hodge, M.R.; Maciejewski, M.; Mu, X.J.; Ra, S.; Zhao, S.; et al. Standard machine learning approaches outperform deep representation learning on phenotype prediction from transcriptomics data. BMC Bioinform. 2020, 21, 119. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning; Springer Series in Statistics; Springer New York: New York, NY, USA, 2009; ISBN 978-0-387-84857-0. [Google Scholar]
- Kobayashi, S.; Saltz, J.H.; Yang, V.W. State of machine and deep learning in histopathological applications in digestive diseases. World J. Gastroenterol. 2021, 27, 2545–2575. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.A.; Ghahramani, Z. Bayesian hierarchical clustering. In Proceedings of the 22nd International Conference on Machine Learning—ICML, Bonn, Germany, 7–11 August 2005; ACM Press: New York, NY, USA, 2005; pp. 297–304. [Google Scholar]
- Abraham, G.; Kowalczyk, A.; Zobel, J.; Inouye, M. Performance and Robustness of Penalized and Unpenalized Methods for Genetic Prediction of Complex Human Disease. Genet. Epidemiol. 2013, 37, 184–195. [Google Scholar] [CrossRef]
- Han, L.; Maciejewski, M.; Brockel, C.; Gordon, W.; Snapper, S.B.; Korzenik, J.R.; Afzelius, L.; Altman, R.B. A probabilistic pathway score (PROPS) for classification with applications to inflammatory bowel disease. Bioinformatics 2018, 34, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Zhang, Y.-H.; Kong, X.-Y.; Cai, Y.-D. Identification of Candidate Genes Related to Inflammatory Bowel Disease Using Minimum Redundancy Maximum Relevance, Incremental Feature Selection, and the Shortest-Path Approach. BioMed Res. Int. 2017, 2017, 5741948. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, D.; Simm, J.; Arany, A.; Fariselli, P.; Cleynen, I.; Moreau, Y. An interpretable low-complexity machine learning framework for robust exome-based in-silico diagnosis of Crohn’s disease patients. NAR Genom. Bioinform. 2020, 2, lqaa011. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Miller, M.; Astrakhan, Y.; Petersen, B.S.; Schreiber, S.; Franke, A.; Bromberg, Y. Identifying Crohn’s disease signal from variome analysis. Genome Med. 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.B.; Lee, S.H.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M.; Gearry, R.B.; Lawrance, I.C.; Andrews, J.M.; Bampton, P.; Mahy, G.; et al. Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med. Genet. 2017, 18, 94. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Wang, W.; Bradfield, J.; Li, J.; Cardinale, C.; Frackelton, E.; Kim, C.; Mentch, F.; Van Steen, K.; Visscher, P.M.; et al. Large sample size, wide variant spectrum, and advanced machine-learning technique boost risk prediction for inflammatory bowel disease. Am. J. Hum. Genet. 2013, 92, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Hübenthal, M.; Hemmrich-Stanisak, G.; Degenhardt, F.; Szymczak, S.; Du, Z.; Elsharawy, A.; Keller, A.; Schreiber, S.; Franke, A. Sparse modeling reveals miRNA signatures for diagnostics of inflammatory bowel disease. PLoS ONE 2015, 10, e0140155. [Google Scholar] [CrossRef] [Green Version]
- Zarringhalam, K.; Enayetallah, A.; Reddy, P.; Ziemek, D. Robust clinical outcome prediction based on Bayesian analysis of transcriptional profiles and prior causal networks. Bioinformatics 2014, 30, i69–i77. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.-S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef]
- Daneshjou, R.; Wang, Y.; Bromberg, Y.; Bovo, S.; Martelli, P.L.; Babbi, G.; Di Lena, P.; Casadio, R.; Edwards, M.; Gifford, D.; et al. Working toward precision medicine: Predicting phenotypes from exomes in the Critical Assessment of Genome Interpretation (CAGI) challenges. Hum. Mutat. 2017, 38, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giollo, M.; Jones, D.T.; Carraro, M.; Leonardi, E.; Ferrari, C.; Tosatto, S.C.E. Crohn disease risk prediction—Best practices and pitfalls with exome data. Hum. Mutat. 2017, 38, 1193–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, L.R.; Yu, C.H.; Mount, S.M.; Moult, J. Insights from GWAS: Emerging landscape of mechanisms underlying complex trait disease. BMC Genom. 2015, 16, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Clyne, M.; Khoury, M.J.; Gwinn, M. Phenopedia and genopedia: Disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 2009, 26, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Barrett, J.C. Genetic risk prediction in complex disease. Hum. Mol. Genet. 2011, 20, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Wray, N.R.; Yang, J.; Goddard, M.E.; Visscher, P.M. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 2010, 6, e1000864. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; Anderson, C.A. Genetic studies of Crohn’s disease: Past, present and future. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Cleynen, I.; González, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlík, M.; Crusius, B.J.A.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleynen, I.; Vermeire, S. The genetic architecture of inflammatory bowel disease: Past, present and future. Curr. Opin. Gastroenterol. 2015, 31, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Nellore, A.; Kammers, K.; Ellis, S.E.; Taub, M.A.; Hansen, K.D.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 2017, 35, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, B.; Dragasevic, S.; Klaassen, K.; Kotur, N.; Srzentic Drazilov, S.; Zukic, B.; Sokic Milutinovic, A.; Milovanovic, T.; Lukic, S.; Popovic, D.; et al. Exploring inflammatory and apoptotic signatures in distinct Crohn’s disease phenotypes: Way towards molecular stratification of patients and targeted therapy. Pathol. Res. Pract. 2020, 216, 152945. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, K.; Roggenbuck, D.; Laass, M.W. Diagnosis and classification of ulcerative colitis. Autoimmun. Rev. 2014, 13, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A blood-based prognostic biomarker in IBD. Gut 2019, 68, 1386–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Zhou, X.; Ma, J.; Zhou, W.; Yang, W.; Fan, D.; Hong, L. Role of MiRNAs in Inflammatory Bowel Disease. Dig. Dis. Sci. 2017, 62, 1426–1438. [Google Scholar] [CrossRef]
- Duttagupta, R.; DiRienzo, S.; Jiang, R.; Bowers, J.; Gollub, J.; Kao, J.; Kearney, K.; Rudolph, D.; Dawany, N.B.; Showe, M.K.; et al. Genome-wide maps of circulating miRNA biomarkers for Ulcerative Colitis. PLoS ONE 2012, 7, e31241. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Hansen, R.; Jones, C.M.A.; Dunn, K.A.; Comeau, A.M.; Bielawski, J.P.; Tayler, R.; El-Omar, E.M.; Russell, R.K.; Hold, G.L.; et al. Multi-omics differentially classify disease state and treatment outcome in pediatric Crohn’s disease. Microbiome 2018, 6, 13. [Google Scholar] [CrossRef]
- Reiman, D.; Layden, B.T.; Dai, Y. MiMeNet: Exploring microbiome-metabolome relationships using neural networks. PLoS Comput. Biol. 2021, 17, e1009021. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.; Simon, J.M.; Kochar, B.; Tovar, A.; Israel, J.W.; Robinson, A.; Gipson, G.R.; Schaner, M.S.; Herfarth, H.H.; Sartor, R.B.; et al. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut 2018, 67, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, R.C.; Hu, L.; Ji, J.; Nayar, S.; Kugathasan, S.; Denson, L.A.; Hyams, J.; Dubinsky, M.C.; Sands, B.E.; Cho, J.H. Machine learning identifies novel blood protein predictors of penetrating and stricturing complications in newly diagnosed paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 2021, 53, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Stafford, I.S.; Kellermann, M.; Mossotto, E.; Beattie, R.M.; MacArthur, B.D.; Ennis, S. A systematic review of the applications of artificial intelligence and machine learning in autoimmune diseases. NPJ Digit. Med. 2020, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Van Der Velde, K.J.; Barbieri, R.; Alberts, R.; Voskuil, M.D.; Vich Vila, A.; Collij, V.; Spekhorst, L.M.; Der Sloot Kwj, V.; Peters, V.; et al. The 1000IBD project: Multi-omics data of 1000 inflammatory bowel disease patients; Data release 1. BMC Gastroenterol. 2019, 19, 5. [Google Scholar] [CrossRef]
- Proctor, L.M.; DiGiacomo, N.D.; Fettweis, J.M.; Jefferson, K.K.; Strauss, J.F.; Rubens, C.E.; Brooks, J.P.; Girerd, P.P.; Huang, B.; Serrano, M.G. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef] [Green Version]



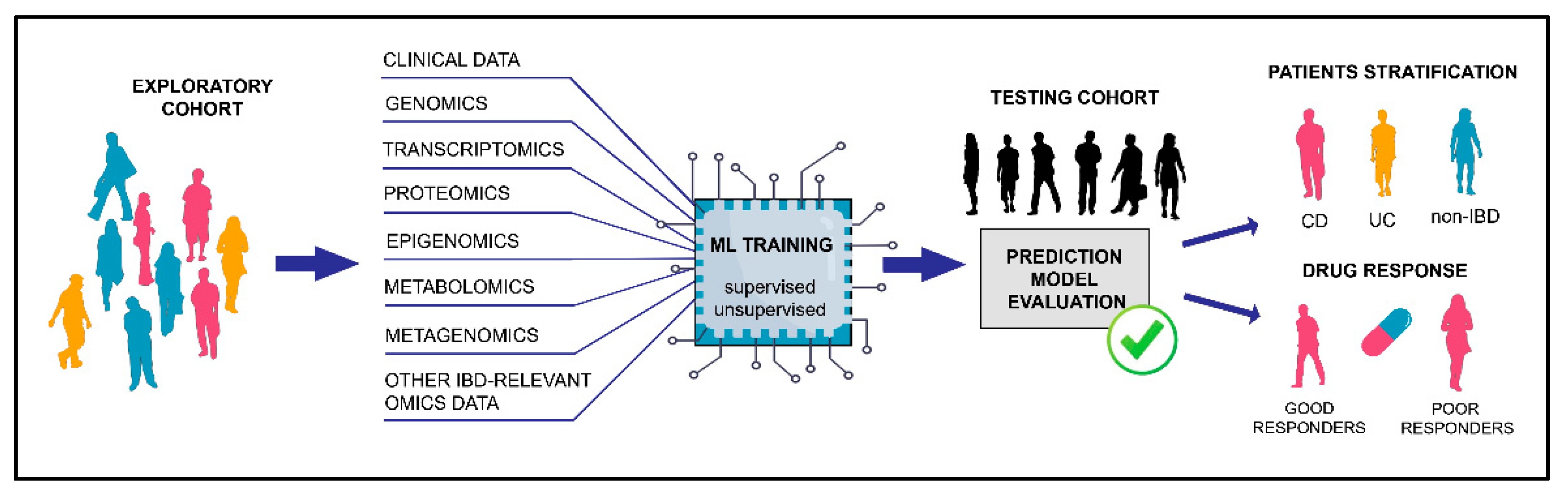{kind=link}
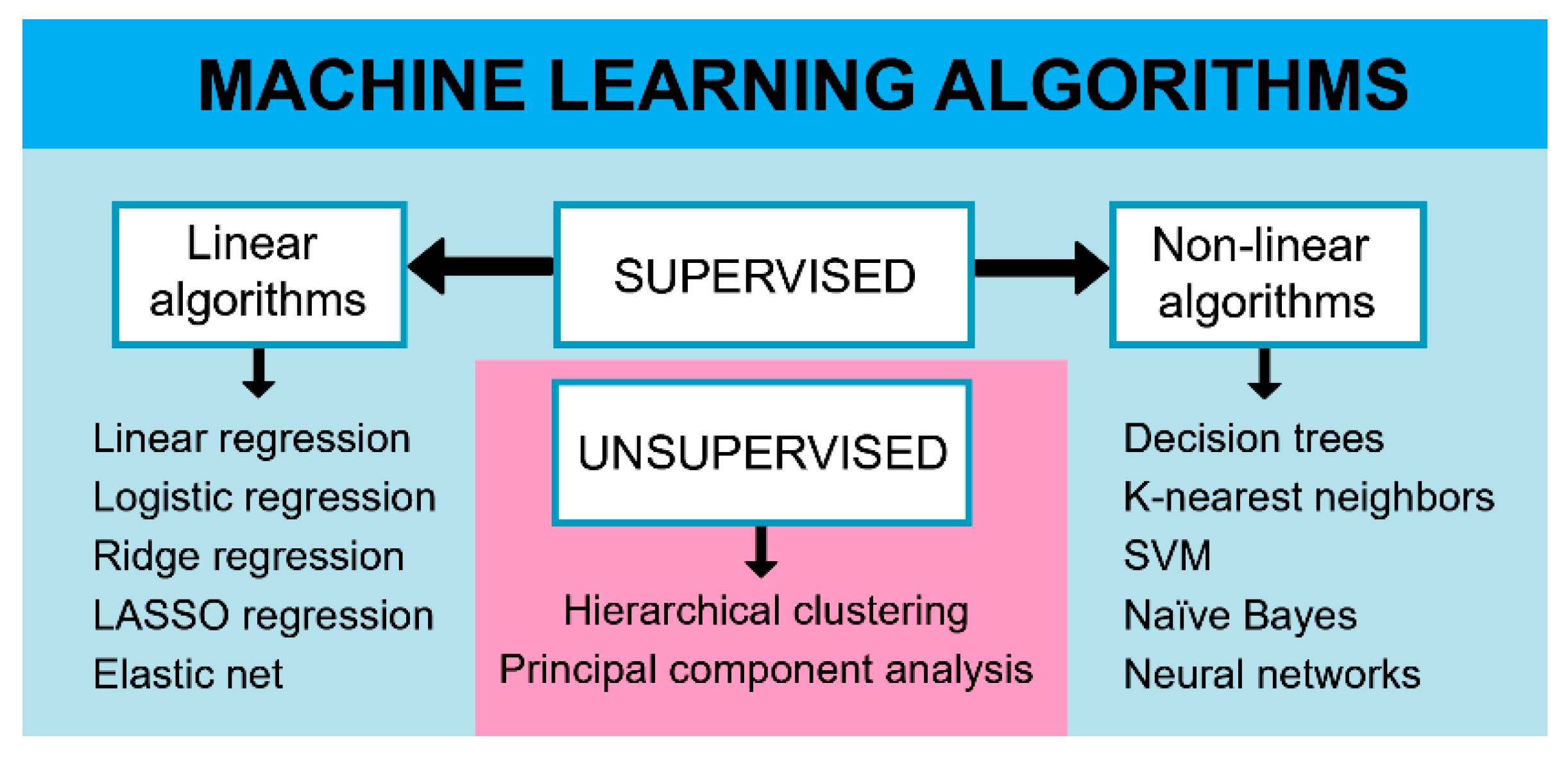{kind=link}
| Instance | An entity (human subject in healthcare applications) which features are used as inputs for prediction modeling. |
| Feature | An explanatory variable, such as genetic variant, gene expression, etc. Features are used as input data for prediction. |
| Machine learning algorithm | Procedure that is run on data to create a machine learning model. It is a set of mathematical optimization functions that minimizes the error of the model function. |
| Iterations | Machine learning algorithm’s parameters are updated number of times until model reaches desired performance |
| Classification | Supervised learning technique used to predict a discrete class or category of an instance (disease or healthy subject, good or poor drug responder, etc.). |
| Regression | Supervised learning technique in which the predicted variable is continuous. |
| Model fitting | Measure of how well a machine learning model generalizes to data not used for model training. |
| Penalized regression method | A method used to reduce overfitting of a model. The penalty causes the regression coefficients of less contributive variables to shrink toward zero therefore reducing the number of variables in the model. |
| Sparse model | A predictive model that includes only the most informative features. |
| Clustering | Unsupervised learning technique that groups instances by their similarity. The groups are called clusters. |
| Black box model | Model that is built on complex functions that are not easily interpreted (such as neural networks). Input and output are clear, but the process between is not explainable. |
| Effect size | A biological measure of the difference or relationship between variables. An OR << 1 or OR >> 1 is indicative of a large effect size. |
| AUC value | Evaluation metric of a model that ranges from 0.5 (poor classifier) to 1 (perfect classifier). |
| Algorithm | Principle | Usage | Pros and Cons |
|---|---|---|---|
| Logistic regression | Linear model transformed into sigmoid function used as a binary classifier | Classification | Fast to develop; easily interpretable; limited by strong assumptions; prone to overfitting |
| Linear regression | Classical linear model that employs linear codependency for prediction | Regression (can also be used for classification) | Fast to develop; easily interpretable; limited by strong assumptions; prone to overfitting |
| Ridge regression | Linear model with L2 regularization | Classification and regression | Linear model with enhanced interpretability and reduced overfitting |
| LASSO | Linear model with L1 regularization | Classification and regression | Linear model with enhanced interpretability and reduced overfitting |
| Elastic net | Linear model with both L1 and L2 regularization | Classification and regression | Linear model with enhanced interpretability and reduced overfitting |
| Decision trees | Prediction based on a tree-like model. Nodes are splitting points of a dataset based on most informative features; leaves are output values. | Classification and regression | Prone to overfitting but can be improved with ensemble methods; interpretable outputs |
| Random forest | An ensemble method (modified bootstrap aggregation) applied to decision trees. It grows multiple decision trees; output is the average prediction of individual trees. | Classification and regression | High prediction performance; deals with overfitting; requires a large dataset for optimal learning. |
| Gradient boosted trees (GBT) | An ensemble method (gradient boosting) applied to decision trees | Classification and regression | High prediction performance; hard-to-tune parameters of the algorithm |
| K nearest neighbors (KNN) | Predicts an output taking into account (k) most similar instances (nearest neighbors) | Classification and regression | Requires a lot of memory to store all the instances; cannot deal with a large number of input variables. |
| Support vector machines (SVM) classifier | Maximizes margin (decision boundary) between different classes supported by instances that lie near the margin (support vectors) | Classification | Works well with high number of input variables; flexible (allow curved margin by using nonlinear kernels); computationally expensive; limited interpretability |
| Naïve Bayes | Employs Bayesian posterior probability theorem but assume nondependency between features given the output | Classification | Fast to develop; suitable for large datasets and for making real time predictions; limited by strong assumptions; requires feature selection and transformation |
| Neural networks | Network of interconnected units resembling the nervous system which renders input information to produce an output. | Classification and regression | High performance; limited interpretability; requires very large dataset; computationally expensive |
| First Author and Year [ref] | Machine Learning Algorithm | Predictors/Prediction | Performance | Tested on Independent Cohort | Subjects |
|---|---|---|---|---|---|
| Chen 2017 [65] | Bayesian mixture approach | GWAS or Immunochip SNPs data/IBD risk score | CD AUC: 0.75, UC AUC: 0.70 | yes | The IIBDGC) cohort—over 68,000 IBD patients and 29,000 healthy controls (4:5 ratio for training and testing, respectively) |
| Wei 2013 [66] | L1 penalized logistic regression, SVM, gradient boosted trees | Immunochip SNPs data/CD and UC distinction from healthy controls | CD AUC 0.86, UC AUC 0.83 | yes | The IIBDGC cohort—~17,000 CD, ~13,000 UC, and ~22,000 controls (randomly divided into 3 folds of equal size for preselection, training and testing, respectively) |
| Romagnoni 2019 [37] | Logistic regression, gradient boosted trees, neural network and ensemble method | Immunochip SNPs data/probability of CD | AUC 0.8 | yes | The IIBDGC cohort—train dataset (34,634 samples), test dataset (17,317 samples) |
| Pal 2017 [51] | Naïve Bayes | Exome data/CD status | AUC 0.81 | yes | Training set: 64 CD and 47 controls (CAGI4); Testing set: 51 CD and 15 controls (CAGI3) |
| Raimondi 2020 [63] | Neural network | Whole exomes/to distinguish between CD and healthy controls | AUC 0.74–0.83 AUPRC 0.81–0.93 | yes | CAGI2, CAGI3, CAGI4 datasets (training and testing) |
| Wang 2019 [64] | SVM | Whole exomes/to distinguish between CD and healthy controls | AUC 0.7–0.75 AUPRC 0.73–0.80 | yes | CAGI4 (training set), CAGI3 (testing set) |
| Isakov 2017 [49] | Random forest, SVM with polynomial kernel, extreme gradient boosting, elastic net and ensemble method | Data from 2050 genes annotated by the expression (array and RNAseq) and pathway information (categorical terms)/IBD-risk gene prioritization | AUC 0.775–0.829 | yes | Intestinal biopsies of 180 CD, 149 UC, 94 colorectal neoplasms, 90 normal tissue (75:25 ratio for training and testing set, respectively) |
| Cushing 2018 [52] | Unsupervised hierarchical clustering, random forest | Whole transcriptome/identification of markers that could predict postoperative disease activity | 92–93% of correct estimates in random forest | no | 24 anti-TNFα-naïve patients, 30 anti-TNFα-exposed |
| Khorasani 2020 [53] | Feature selection algorithm (based on dimension reduction) combined with SVM classifier | Wide expression array data/UC and healthy subjects classification | Active UC AUPRC 1, Inactive UC AUPRC 0.68 | yes | Training set: 39 UC samples (active and inactive) and 38 controls; testing set: 97 UC samples (active and inactive) and 22 controls |
| Yuan 2017 [62] | Feature selection (minimum redundancy maximum relevance and incremental feature selection), SVM-based algorithm (sequential minimal optimization) | Wide expression array data from PBMC samples/CD, UC and normal subject discrimination and candidate gene selection | Accuracy 0.94 | no | 59 Crohn’s disease, 26 ulcerative colitis, and 42 normal samples |
| Hubenthal 2015 [67] | Penalized SVM, random forest | miRNAs in whole-blood samples/IBD and control subject distinction | AUC 0.75-1.0 | no | 40 CD, 36 UC, 38 healthy controls and other inflammation controls (24 chronic obstructive pulmonary disease, 23 multiple sclerosis, 38 pancreatitis and 45 sarcoidosis cases) |
| Zarringhalam 2014 [68] | Differential expression profile was used to infer upstream regulators using Bayesian approach; posterior probabilities of regulators’ activities were then used in a regularized regression framework to predict outcome | Genome wide expression data/response to infliximab in UC | Accuracy 0.79 | yes | Training set: 22 active UC patients (12 responders and 10 nonresponders); Testing set: 24 active UC patients (8 responders and 16 nonresponders) |
| Li 2020 [50] | Random forest, neural network | RNAseq and microarray expression data/identification of susceptibility genes and establishing predictive model of UC | AUC 0.95; AUPRC 0.97 | yes | Training set: 206 UC, 20 normal; Testing set: 53 UC and 21 normal |
| Martin 2019 [69] | Hierarchical clustering, principal component analysis | Single-cell RNA sequencing data/cell type classification in inflamed and uninflamed tissues | Inflamed tissue (r = 0.96) Uninflamed tissue (r = 0.93) * | no | 11 ileal CD patients; samples taken from inflamed and uninflamed tissues |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stankovic, B.; Kotur, N.; Nikcevic, G.; Gasic, V.; Zukic, B.; Pavlovic, S. Machine Learning Modeling from Omics Data as Prospective Tool for Improvement of Inflammatory Bowel Disease Diagnosis and Clinical Classifications. Genes 2021, 12, 1438. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091438
Stankovic B, Kotur N, Nikcevic G, Gasic V, Zukic B, Pavlovic S. Machine Learning Modeling from Omics Data as Prospective Tool for Improvement of Inflammatory Bowel Disease Diagnosis and Clinical Classifications. Genes. 2021; 12(9):1438. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091438
Chicago/Turabian StyleStankovic, Biljana, Nikola Kotur, Gordana Nikcevic, Vladimir Gasic, Branka Zukic, and Sonja Pavlovic. 2021. "Machine Learning Modeling from Omics Data as Prospective Tool for Improvement of Inflammatory Bowel Disease Diagnosis and Clinical Classifications" Genes 12, no. 9: 1438. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091438