
ORPER: A Workflow for Constrained SSU rRNA Phylogenies


1
BCCM/IHEM, Mycology and Aerobiology, Sciensano, 1050 Bruxelles, Belgium
2
BCCM/ULC Collection, InBioS–Centre for Protein Engineering, University of Liège, 4000 Liège, Belgium
3
InBioS–PhytoSYSTEMS, Unit of Eukaryotic Phylogenomics, University of Liège, 4000 Liège, Belgium
*
Authors to whom correspondence should be addressed.
Genes 2021, 12(11), 1741; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111741
Submission received: 5 October 2021
/
Revised: 24 October 2021
/
Accepted: 28 October 2021
/
Published: 29 October 2021
(This article belongs to the Special Issue Genomics, Metagenomics and Phylogenomics of Cyanobacteria and Related Lineages)
{kind=link}
{kind=link}
Abstract
:The continuous increase in sequenced genomes in public repositories makes the choice of interesting bacterial strains for future sequencing projects ever more complicated, as it is difficult to estimate the redundancy between these strains and the already available genomes. Therefore, we developed the Nextflow workflow “ORPER”, for “ORganism PlacER”, containerized in Singularity, which allows the determination the phylogenetic position of a collection of organisms in the genomic landscape. ORPER constrains the phylogenetic placement of SSU (16S) rRNA sequences in a multilocus reference tree based on ribosomal protein genes extracted from public genomes. We demonstrate the utility of ORPER on the Cyanobacteria phylum, by placing 152 strains of the BCCM/ULC collection.
1. Introduction
Cyanobacteria form a phylum of bacteria, which have colonized very diversified ecosystems [1]. They are the only bacteria able to perform oxygenic photosynthesis and appeared at least 2.4 billion years ago [2]. By increasing the free atmospheric oxygen, Cyanobacteria had a critical impact on shaping life on Earth [3,4]. Beyond their ecological importance, this phylum also has an evolutionary interest due to their key role in the emergence of Archaeplastida through the primary endosymbiosis, which gave rise to the plastid [5]. Although the exact mechanisms, which include the generally accepted unicity of the event, are yet to be fully understood, it is well known that Cyanobacteria played a major role in the spread of oxygenic photosynthesis [6]. More recently, the group attracted an additional interest after uncovering, through metagenomic studies, the existence of non-photosynthetic “cyanobacteria”, notably the phylum Melainabacteria [7].
Due to this importance, the published cyanobacterial phylogenies are numerous (see for instance: [8,9,10,11,12,13,14]). The number of available genomes logically followed this interest, rising from a few hundred in 2013, when Shih et al. [15] improved the coverage of the phylum, to more than 3000 nowadays, according to GenBank statistics. Nevertheless, recent studies have demonstrated that cyanobacterial diversity, both for photosynthetic [16] and non-photosynthetic [14] representatives (when considering Melainabacteria as part of Cyanobacteria), is not well covered by the sequencing effort.
The gold standard for the estimation of bacterial diversity remains the SSU rRNA gene of the small subunit of the ribosomal RNA [17]. This locus is frequently used by scientists and culture collections to evaluate the genomic potential of newly isolated organisms. However, due to the constant and rapid growth of genome repositories, it is difficult for researchers to estimate the redundancy between these public data sources and their own collections of organisms. Here, we release ORPER, which stands for “ORganism PlacER”, an automated workflow intended to determine the phylogenetic position of organisms, for which only the SSU rRNA has been determined, in the public genomic landscape.
2. Methods
2.1. Functional Overview
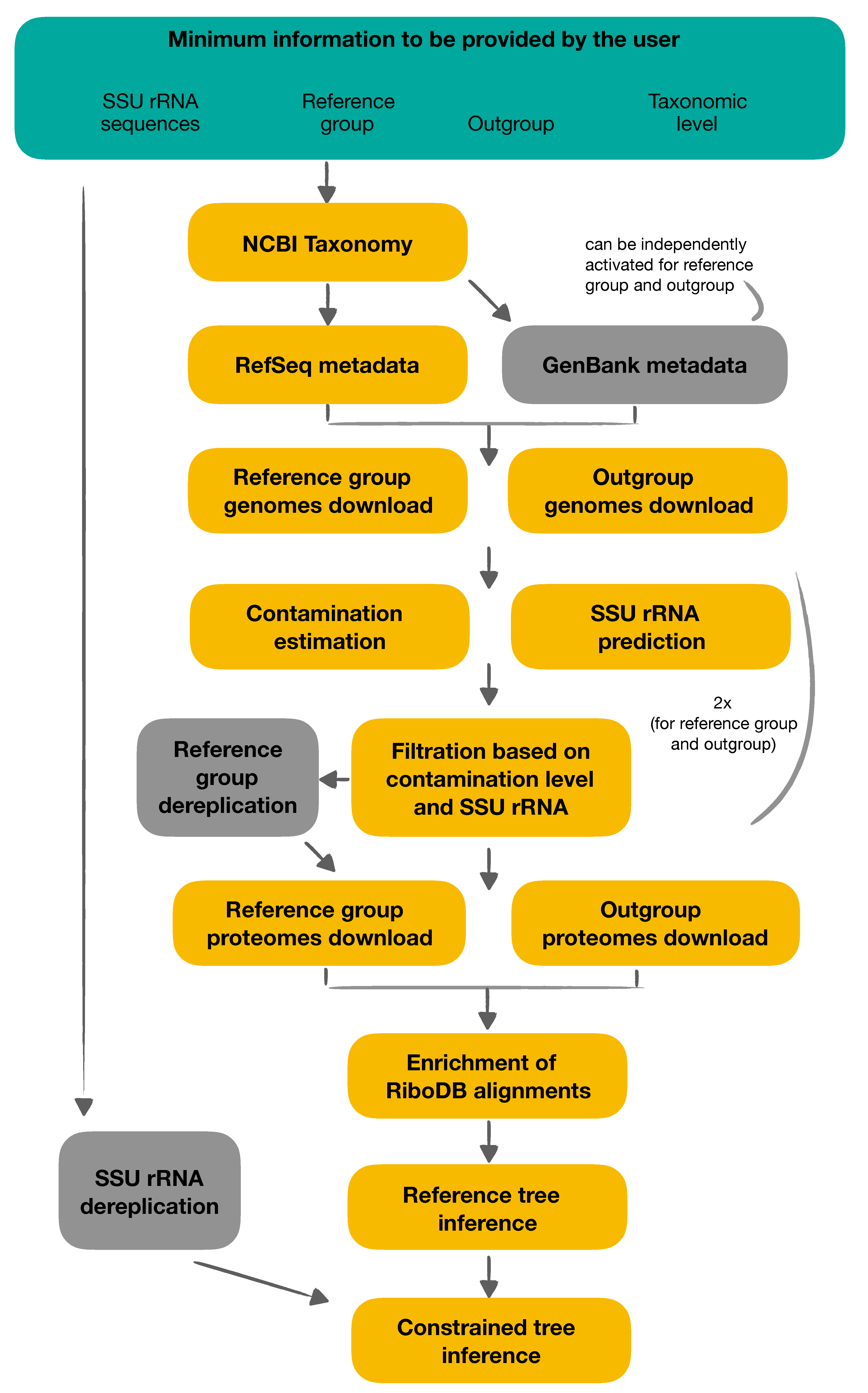
The principle of ORPER is to provide an overview of the sequenced coverage (i.e., the diversity of available genomes) of a given taxon and to place SSU rRNA sequences in this diversity. ORPER first downloads the complete genomes of the taxon of interest, then extracts their ribosomal proteins to compute a reference phylogenetic tree, and finally uses this tree to constrain the backbone of a SSU rRNA phylogeny including the additional strains. The workflow uses two groups: (i) the main group corresponding to the taxonomic group of the SSU rRNA sequences (the taxon of interest) and (ii) the outgroup to the root of the phylogenetic tree. The main group is used to compute a phylogenetic tree to guide the placement of SSU rRNA sequences; therefore, it is named “reference group” for the remainder of this manuscript (Figure 1). All steps are embedded in a Nextflow script [18], and a Singularity definition file is provided for containerization [19]. ORPER is available at https://github.com/Lcornet/ORPER, accessed on 1 October 2021.
2.2. Workflow Details
2.2.1. Taxonomy and Metadata Download
ORPER begins by creating a local copy of the National Center for Biotechnology Information (NCBI) Taxonomy [20] with the script setup-taxdir.pl v0.211470 from Bio::MUST::Core (D. Baurain; https://metacpan.org/dist/Bio-MUST-Core, accessed on 1 October 2021. Genome accession numbers (i.e., GCF numbers) are fetched from the NCBI Reference Sequence project (RefSeq) [21] and the taxonomy of the corresponding organisms is determined with the script fetch-tax.pl v0.211470 (Bio::MUST::Core package). If required, GenBank genomes [22] can be used in the same way for the both reference group and outgroup creation, independently. Four taxonomic levels are available in ORPER (phylum, class, order, family) and the user must specify the reference group and the outgroup separately (Figure 1).
2.2.2. Genome Filtration and Dereplication
CheckM v1.1.3 with the “lineage_wf” option, is used to estimate completeness and contamination of the assemblies [23]. Barrnap v0.9, with default options, is used to predict rRNA genes in downloaded genomes (available at https://github.com/tseemann/barrnap, accessed on 1 October 2021). Genomes with a completeness level above 90%, a contamination level below 5%, and at least one predicted SSU rRNA sequence are retained. A dereplication step of the genomes from the reference group can be optionally carried out using dRep [24] and default parameters. Prodigal, with default options, is used to obtain conceptual proteomes [25]. All genomes from the reference group that remain after the filtration steps are used, whereas only the ten first genomes of the outgroup are used for de novo protein prediction (Figure 1).
2.2.3. Reference Phylogeny Inference
Prokaryotic ribosomal protein alignments from the RiboDB database [26] are downloaded by ORPER once at the first usage. An orthologous enrichment of these alignments with sequences from the remaining proteomes (post-filtration and dereplication) is performed by Forty-Two v0.210570 [27,28]. These sequences are then aligned using MUSCLE v3.8.31 [29] in order to generate new alignment files with only the sequences from the reference group and the outgroup. Conserved sites are selected using BMGE v1.12 [30] with moderately severe settings (entropy cut-off = 0.5, gap cut-off = 0.2). A supermatrix is then generated using SCaFoS v1.30k [31] with default settings. Finally, a reference phylogenomic analysis is inferred using RAxML v8.2.12 [32] with 100 bootstrap replicates under the PROTGAMMALGF model.
2.2.4. Constrained SSU rRNA Phylogeny
The SSU rRNA sequences provided by the user can be optionally dereplicated using CD-HIT-EST v4.8.1 with default parameters [33]. The SSU rRNA phylogenetic tree is inferred from both the sequences provided by the user and those extracted from the complete genomes using RAxML v8.2.12 [32] with 100 bootstrap replicates under the GTRGAMMA model and the phylogenomic tree as a constraint.
2.3. Design Considerations
ORPER compensates for the lack of phylogenetic resolution of SSU rRNA gene sequences by using ribosomal protein genes from publicly available genomes to infer a reference multilocus tree, which is then used to constrain the SSU rRNA phylogeny. Indeed, it is well known that SSU rRNA suffers, as do all single-gene phylogenies, from a lack of phylogenetic resolution [34,35,36,37,38]. Ribosomal protein genes are frequently used to perform phylogenetic placement; for instance, CheckM uses this approach to place genomes before performing the contamination estimation [23].
The NCBI databases, regularly synchronized with the European Nucleotide Archive (ENA) [39], are the most complete public databases. By default, ORPER uses only RefSeq because the latter contains only high-quality genomes [21]. Nevertheless, it might be necessary to use more genomes to estimate the actual sequence coverage of a taxon. This is especially true with metagenomic data that are, by design, not included in RefSeq [21]. That is why GenBank can be enabled as an option in ORPER. In any case, starting from a NCBI database entails the use of thousands of genomes, which can dramatically increase the computing time. For this reason, we implemented the optional use of dRep to dereplicate the genomes [24]. This allowed the user to decrease the number of genomes while conserving the sequenced diversity [24]. However, this option should be used carefully because the need for dereplication (or not) is dependent on the biological question [40]. For example, the genomic comparison of closely related strains requires using as many genomes as possible to identify individual differences. Finally, genomes in public repositories are not devoid of contamination (i.e., the inclusion of foreign DNA in the genomic data) [41,42]. Therefore, we used CheckM [23], the most commonly used tool for genomic contamination detection, and thresholds from the Genomic Standards Consortium [43] (completeness above 90% and contamination below 5%) to filter our genomes, which is a mandatory step in the workflow.
Nextflow is the latest workflow system. It was developed to increase reproducibility in science [18]. Nextflow further presents the advantage of exploiting Singularity containers as an operating system [18], which ensures the sustainability of future analyses. Singularity containers [19] correct the security issues of older container systems, thereby offering the possibility of deploying them on HPC systems where security is often an important concern. Owing to these advantages, we chose the combination Nextflow-Singularity for ORPER. Albeit ORPER is a workflow, we designed it as a program with a single command-line interface. The installation of ORPER only requires two shell commands (see https://github.com/Lcornet/ORPER, accessed on 1 October 2021). Moreover, the analysis reported in this study can be replayed with a single command in less than one day using 30 CPU cores (Intel Xeon E5-2640 v4 series) (see https://github.com/Lcornet/ORPER, accessed on 1 October 2021).
3. Results and Discussion
Case Study: BCCM/ULC Cyanobacteria Collection
Phylogenomic studies of Cyanobacteria are numerous, notably focusing on the emergence of multicellularity [44,45,46], the appearance of oxygenic photosynthesis [47,48,49], or the origin of plastids [9,50,51,52]. The emergence of the plastid remains quite unclear [6] with potential origins either among heterocyst-forming cyanobacteria [2,53] or earlier diverging lineages [9,50,51,52]. Therefore, the selection of Cyanobacteria for future sequencing projects remains an important issue.
We tested ORPER on this phylum with 152 SSU rRNA sequences from the BCCM/ULC collection. The information on the SSU rRNA sequences and the collection itself is available in Supplementary File S1. RefSeq genomes for the “Cyanobacteria” phylum were specified as the reference group, whereas genomes for the “Melainabacteria” phylum available in GenBank were used as the outgroup (Figure 2). The dereplications for the reference genomes and for the SSU rRNA sequences were both activated. The reference tree inferred by ORPER was based on a supermatrix of 372 organisms × 6246 unambiguously aligned amino-acid positions (7.92% missing character states). The 152 SSU rRNA input sequences used in this study were dereplicated to 140 sequences at a 95% identity threshold, and were then used to compute the constrained tree.
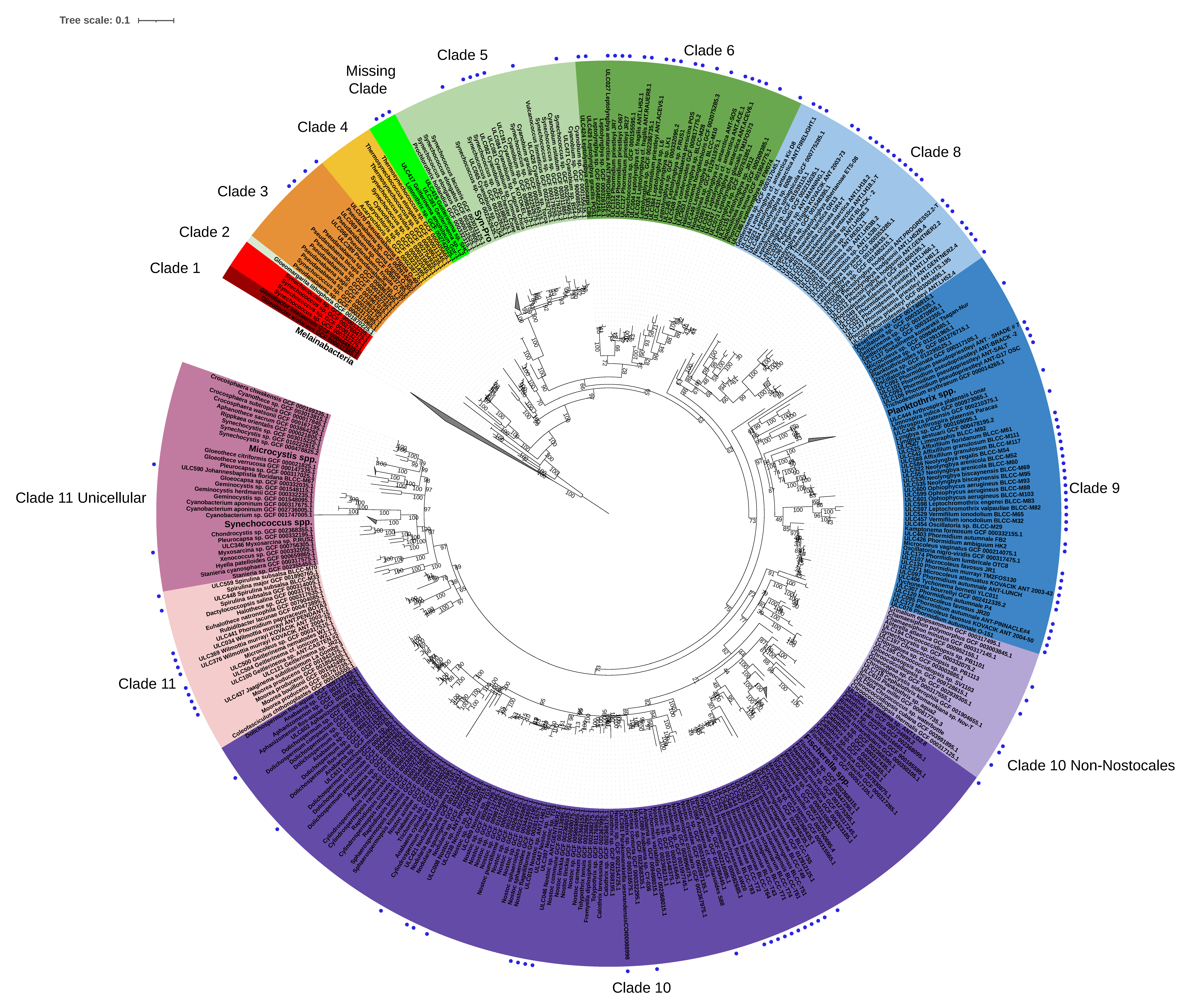
We chose to compare the phylogeny inferred by ORPER to the latest multilocus (ribosomal) phylogeny of the cyanobacterial phylum published by Moore et al. (2019), who identified the earliest potential basal position of the plastids [9]. The constrained tree computed by ORPER is comparable to the tree of Moore et al. (2019), with ten out of eleven clades recovered by ORPER (Figure 2). The only missing clade, clade 7, was represented by genomes neither present in RefSeq [9] nor in the ULC strains, and thus was logically absent from our phylogeny. The 140 BCCM/ULC strains obtained after dereplication covered the whole diversity of publicly available cyanobacterial genomes. Three BCCM/ULC strains (ULC415, ULC417, ULC381) formed a basal clade clustered with Limnothrix sp. GCF_002742025.1, which was not present in Moore et al.’s analysis (Supplementary File S2). These three strains are, therefore, of high interest for genome sequencing, especially in the context of plastid emergence. Here, we analyzed the cyanobacterial phylum, but ORPER could be used on any bacterial taxon of the NCBI.
4. Conclusions
ORPER is a state-of-the-art tool, designed for the phylogenetic placement of SSU rRNA sequences in a phylogenetic tree constrained by a multilocus tree. We demonstrated the utility of ORPER on Cyanobacteria, using sequences from the BCCM/ULC collection, to estimate the phylogenetic position of SSU rRNA sequences among the landscape of sequenced genomes. Its easy-to-use installation process and Singularity containerization makes ORPER a useful tool for culture collections and for scientists to use in their future selection of genomes to sequence.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/genes12111741/s1, Supplementary file S1: Information on the 152 ULC strains used in this study. Supplementary file S2: Vertical representation of the phylogenetic tree shown in Figure 2.
Author Contributions
L.C., D.B. conceived the study. L.C. performed all the analyses and drew the figures. A.-C.A. provided the SSU rRNA gene sequences of the BCCM/ULC strains and their information. L.C. and D.B. drafted the manuscript, and A.-C.A. and A.W. provided critical reviews of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a research grant (no. B2/191/P2/BCCM GEN-ERA) funded by the Belgian Science Policy Office (BELSPO). Computational resources were provided by the Consortium des Équipements de Calcul Intensif (CÉCI) funded by the F.R.S.-FNRS (2.5020.11), and through two research grants to DB: B2/191/P2/BCCM GEN-ERA (Belgian Science Policy Office—BELSPO) and CDR J.0008.20 (F.R.S.-FNRS). A.W. is a Senior Research Associate of the FRS-FNRS. The BCCM/ULC collection is supported by the Belgian Science Policy Office (C5/00/BCCM).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
ORPER is freely available at https://github.com/Lcornet/ORPER (accessed on 1 October 2021).
Acknowledgments
The authors thank David Colignon (ULiège) and the CÉCI for their help with computing cluster usage.
Conflicts of Interest
The authors declare that there are no conflict of interest.
References
- Whitton, B.A. Ecology of Cyanobacteria II: Their Diversity in Space and Time; Springer Science & Business Media: Dordrecht, The Netherlands, 2012; ISBN 978-94-007-3855-3. [Google Scholar]
- Ochoa de Alda, J.A.G.; Esteban, R.; Diago, M.L.; Houmard, J. The Plastid Ancestor Originated among One of the Major Cyanobacterial Lineages. Nat. Commun. 2014, 5, 4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, R.E.; Kirschvink, J.L.; Hilburn, I.A.; Nash, C.Z. The Paleoproterozoic Snowball Earth: A Climate Disaster Triggered by the Evolution of Oxygenic Photosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 11131–11136. [Google Scholar] [CrossRef] [Green Version]
- Knoll, A.H. The Geological Consequences of Evolution. Geobiology 2003, 1, 3–14. [Google Scholar] [CrossRef]
- Archibald, J.M. The Puzzle of Plastid Evolution. Curr. Biol. 2009, 19, R81–R88. [Google Scholar] [CrossRef] [Green Version]
- Sato, N. Are Cyanobacteria an Ancestor of Chloroplasts or Just One of the Gene Donors for Plants and Algae? Genes 2021, 12, 823. [Google Scholar] [CrossRef]
- Di Rienzi, S.C.; Sharon, I.; Wrighton, K.C.; Koren, O.; Hug, L.A.; Thomas, B.C.; Goodrich, J.K.; Bell, J.T.; Spector, T.D.; Banfield, J.F.; et al. The Human Gut and Groundwater Harbor Non-Photosynthetic Bacteria Belonging to a New Candidate Phylum Sibling to Cyanobacteria. eLife 2013, 2, e01102. [Google Scholar] [CrossRef]
- Mareš, J.; Strunecký, O.; Bučinská, L.; Wiedermannová, J. Evolutionary Patterns of Thylakoid Architecture in Cyanobacteria. Front. Microbiol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.R.; Magnabosco, C.; Momper, L.; Gold, D.A.; Bosak, T.; Fournier, G.P. An Expanded Ribosomal Phylogeny of Cyanobacteria Supports a Deep Placement of Plastids. Front. Microbiol. 2019, 10, 1612. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Baracaldo, P.; Cardona, T. On the Origin of Oxygenic Photosynthesis and Cyanobacteria. New Phytol. 2020, 225, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Baracaldo, P.; Bianchini, G.; Wilson, J.D.; Knoll, A.H. Cyanobacteria and Biogeochemical Cycles through Earth History. Trends Microbiol. 2021. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Teng, W.-K.; Zhao, L.; Hu, C.-X.; Zhou, Y.-K.; Han, B.-P.; Song, L.-R.; Shu, W.-S. Comparative Genomics Reveals Insights into Cyanobacterial Evolution and Habitat Adaptation. ISME J. 2021, 15, 211–227. [Google Scholar] [CrossRef]
- Boden, J.S.; Konhauser, K.O.; Robbins, L.J.; Sánchez-Baracaldo, P. Timing the Evolution of Antioxidant Enzymes in Cyanobacteria. Nat. Commun. 2021, 12, 4742. [Google Scholar] [CrossRef]
- Monchamp, M.-E.; Spaak, P.; Pomati, F. Long Term Diversity and Distribution of Non-Photosynthetic Cyanobacteria in Peri-Alpine Lakes. Front. Microbiol. 2019, 9, 3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; de Marsac, N.T.; Rippka, R.; et al. Improving the Coverage of the Cyanobacterial Phylum Using Diversity-Driven Genome Sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornet, L.; Wilmotte, A.; Javaux, E.J.; Baurain, D. A Constrained SSU-rRNA Phylogeny Reveals the Unsequenced Diversity of Photosynthetic Cyanobacteria (Oxyphotobacteria). BMC Res. Notes 2018, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020. [Google Scholar] [CrossRef]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An Update on Prokaryotic Genome Annotation and Curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. DRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauffrit, F.; Penel, S.; Delmotte, S.; Rey, C.; de Vienne, D.M.; Gouy, M.; Charrier, J.-P.; Flandrois, J.-P.; Brochier-Armanet, C. RiboDB Database: A Comprehensive Resource for Prokaryotic Systematics. Mol. Biol. Evol. 2016, 33, 2170–2172. [Google Scholar] [CrossRef] [Green Version]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D.J.; Di Franco, A.; Roure, B.; Satoh, N.; Quéinnec, É.; Ereskovsky, A.; et al. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Curr. Biol. 2017, 27, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, M.; Di Franco, A.; Philippe, H.; Baurain, D. Decontamination, Pooling and Dereplication of the 678 Samples of the Marine Microbial Eukaryote Transcriptome Sequencing Project. BMC Res. Notes 2021, 14, 306. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A New Software for Selection of Phylogenetic Informative Regions from Multiple Sequence Alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Roure, B.; Rodriguez-Ezpeleta, N.; Philippe, H. SCaFoS: A Tool for Selection, Concatenation and Fusion of Sequences for Phylogenomics. BMC Evol. Biol. 2007, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum Likelihood-Based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Gontcharov, A.A.; Marin, B.; Melkonian, M. Are Combined Analyses Better Than Single Gene Phylogenies? A Case Study Using SSU RDNA and RbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2004, 21, 612–624. [Google Scholar] [CrossRef]
- Dessimoz, C.; Gil, M. Phylogenetic Assessment of Alignments Reveals Neglected Tree Signal in Gaps. Genome Biol. 2010, 11, R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunter, G.; Rocco, A.; Mimouni, N.; Heger, A.; Caldeira, A.; Hein, J. Uncertainty in Homology Inferences: Assessing and Improving Genomic Sequence Alignment. Genome Res. 2008, 18, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.M.; Suchard, M.A.; Huelsenbeck, J.P. Alignment Uncertainty and Genomic Analysis. Science 2008, 319, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareš, J. Multilocus and SSU rRNA Gene Phylogenetic Analyses of Available Cyanobacterial Genomes, and Their Relation to the Current Taxonomic System. Hydrobiologia 2018, 811, 19–34. [Google Scholar] [CrossRef]
- Harrison, P.W.; Ahamed, A.; Aslam, R.; Alako, B.T.F.; Burgin, J.; Buso, N.; Courtot, M.; Fan, J.; Gupta, D.; Haseeb, M.; et al. The European Nucleotide Archive in 2020. Nucleic Acids Res. 2021, 49, D82–D85. [Google Scholar] [CrossRef]
- Evans, J.T.; Denef, V.J. To Dereplicate or Not to Dereplicate? mSphere 2020, 5, e00971-19. [Google Scholar] [CrossRef]
- Cornet, L.; Meunier, L.; Vlierberghe, M.V.; Léonard, R.R.; Durieu, B.; Lara, Y.; Misztak, A.; Sirjacobs, D.; Javaux, E.J.; Philippe, H.; et al. Consensus Assessment of the Contamination Level of Publicly Available Cyanobacterial Genomes. PLoS ONE 2018, 13, e0200323. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Pertea, M.; Zimin, A.V.; Salzberg, S.L. Human Contamination in Bacterial Genomes Has Created Thousands of Spurious Proteins. Genome Res. 2019, 29, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, B.E.; Antonelli, A.; Bagheri, H.C. The Origin of Multicellularity in Cyanobacteria. BMC Evol. Biol. 2011, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, B.E.; de Vos, J.M.; Antonelli, A.; Bagheri, H.C. Evolution of Multicellularity Coincided with Increased Diversification of Cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 2013, 110, 1791–1796. [Google Scholar] [CrossRef] [Green Version]
- Urrejola, C.; von Dassow, P.; van den Engh, G.; Salas, L.; Mullineaux, C.W.; Vicuña, R.; Sánchez-Baracaldo, P. Loss of Filamentous Multicellularity in Cyanobacteria: The Extremophile Gloeocapsopsis Sp. Strain UTEX B3054 Retained Multicellular Features at the Genomic and Behavioral Levels. J. Bacteriol. 2021, 2021, e00514-19. [Google Scholar] [CrossRef]
- Oliver, T.; Sánchez-Baracaldo, P.; Larkum, A.W.; Rutherford, A.W.; Cardona, T. Time-Resolved Comparative Molecular Evolution of Oxygenic Photosynthesis. Biochim. Et Biophys. Acta BBA Bioenerg. 2021, 1862, 148400. [Google Scholar] [CrossRef]
- Cardona, T. Thinking Twice about the Evolution of Photosynthesis. Open Biol. 2019, 2019, 180246. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pichel, F.; Lombard, J.; Soule, T.; Dunaj, S.; Wu, S.H.; Wojciechowski, M.F. Timing the Evolutionary Advent of Cyanobacteria and the Later Great Oxidation Event Using Gene Phylogenies of a Sunscreen. mBio 2021, 10, e00561-19. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Toledo, R.I.; Deschamps, P.; López-García, P.; Zivanovic, Y.; Benzerara, K.; Moreira, D. An Early-Branching Freshwater Cyanobacterium at the Origin of Plastids. Curr. Biol. 2017, 27, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce-Toledo, R.I.; López-García, P.; Moreira, D. Horizontal and Endosymbiotic Gene Transfer in Early Plastid Evolution. New Phytol. 2019, 224, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. Large-Scale Phylogenomic Analyses Indicate a Deep Origin of Primary Plastids within Cyanobacteria. Mol. Biol. Evol. 2011, 28, 3019–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, O.; Landan, G.; Roettger, M.; Gruenheit, N.; Kowallik, K.V.; Allen, J.F.; Martin, W.; Dagan, T. Genes of Cyanobacterial Origin in Plant Nuclear Genomes Point to a Heterocyst-Forming Plastid Ancestor. Mol. Biol. Evol. 2008, 25, 748–761. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of ORPER workflow. Users should specify at least four pieces of information to run ORPER: (i) their SSU (16S) rRNA sequences, (ii) the taxon of interest, (iii) the outgroup of the phylogeny and (iv) the taxonomic level (Green part). Yellow boxes are mandatory steps of ORPER whereas grey boxes are optional steps. Contamination estimation, SSU rRNA prediction and filtration are performed twice, once for the reference group and once for the outgroup.
Figure 1.
Overview of ORPER workflow. Users should specify at least four pieces of information to run ORPER: (i) their SSU (16S) rRNA sequences, (ii) the taxon of interest, (iii) the outgroup of the phylogeny and (iv) the taxonomic level (Green part). Yellow boxes are mandatory steps of ORPER whereas grey boxes are optional steps. Contamination estimation, SSU rRNA prediction and filtration are performed twice, once for the reference group and once for the outgroup.

Figure 2.
Constrained cyanobacterial phylogenetic tree of the BCCM/ULC collection. The tree is the output of ORPER, a Maximum-likelihood constrained inference computed under the GTRGAMMA model. Clades correspond to the groups defined in Moore et al. (2019) [9]. Clades 10 and 11 have been divided into two sub-clades, adding, respectively “Non-Nostocales” and “Unicellular” sub-clades to Moore et al.’s phylogeny. Blue dots indicate ULC/BCCM strains. The clade absent from Moore et al.’s phylogeny is indicated as “Missing Clade”.
Figure 2.
Constrained cyanobacterial phylogenetic tree of the BCCM/ULC collection. The tree is the output of ORPER, a Maximum-likelihood constrained inference computed under the GTRGAMMA model. Clades correspond to the groups defined in Moore et al. (2019) [9]. Clades 10 and 11 have been divided into two sub-clades, adding, respectively “Non-Nostocales” and “Unicellular” sub-clades to Moore et al.’s phylogeny. Blue dots indicate ULC/BCCM strains. The clade absent from Moore et al.’s phylogeny is indicated as “Missing Clade”.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cornet, L.; Ahn, A.-C.; Wilmotte, A.; Baurain, D. ORPER: A Workflow for Constrained SSU rRNA Phylogenies. Genes 2021, 12, 1741. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111741
AMA Style
Cornet L, Ahn A-C, Wilmotte A, Baurain D. ORPER: A Workflow for Constrained SSU rRNA Phylogenies. Genes. 2021; 12(11):1741. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111741
Chicago/Turabian StyleCornet, Luc, Anne-Catherine Ahn, Annick Wilmotte, and Denis Baurain. 2021. "ORPER: A Workflow for Constrained SSU rRNA Phylogenies" Genes 12, no. 11: 1741. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12111741
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.