
Whole Genome Sequencing Unravels New Genetic Determinants of Early-Onset Familial Osteoporosis and Low BMD in Malta

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Compliance
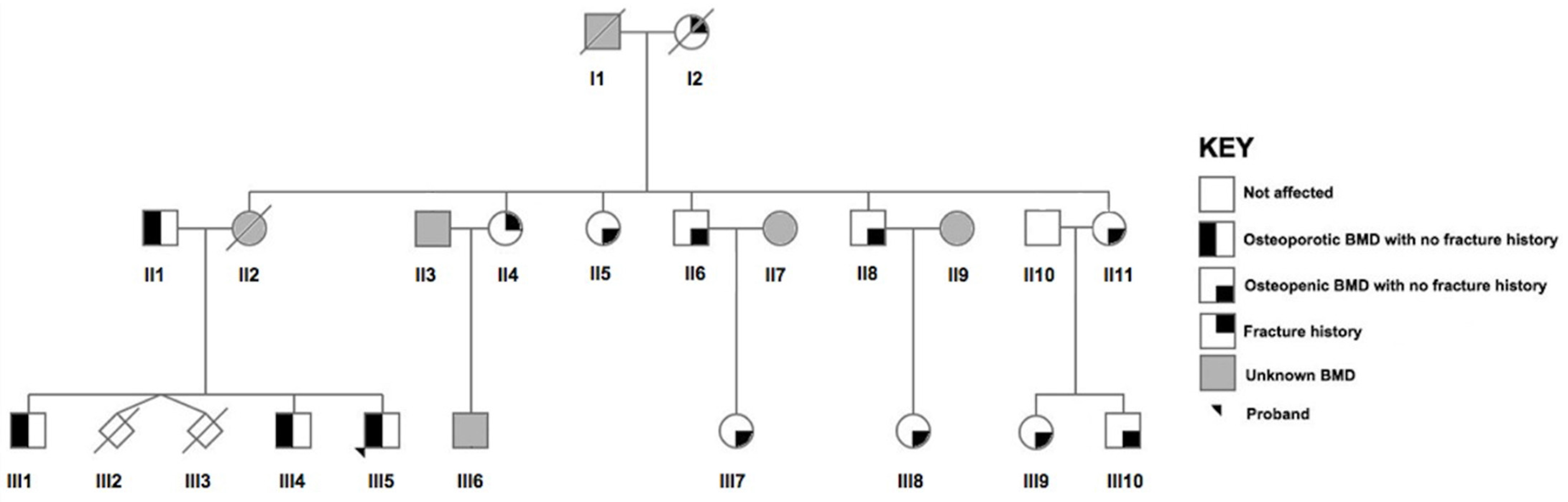
2.2. Recruitment of the Maltese Pedigree
2.3. BMD Measurements and Phenotype Definition
2.4. Collection of Anthropometric Data and Biological Samples
2.5. DNA Extraction and WGS
2.6. Bioinformatics Processing and Data Analysis
2.7. Genotyping of Shortlisted Variants in a Maltese Population Study
2.8. Statistical Analysis
3. Results
3.1. WGS Data Filtering
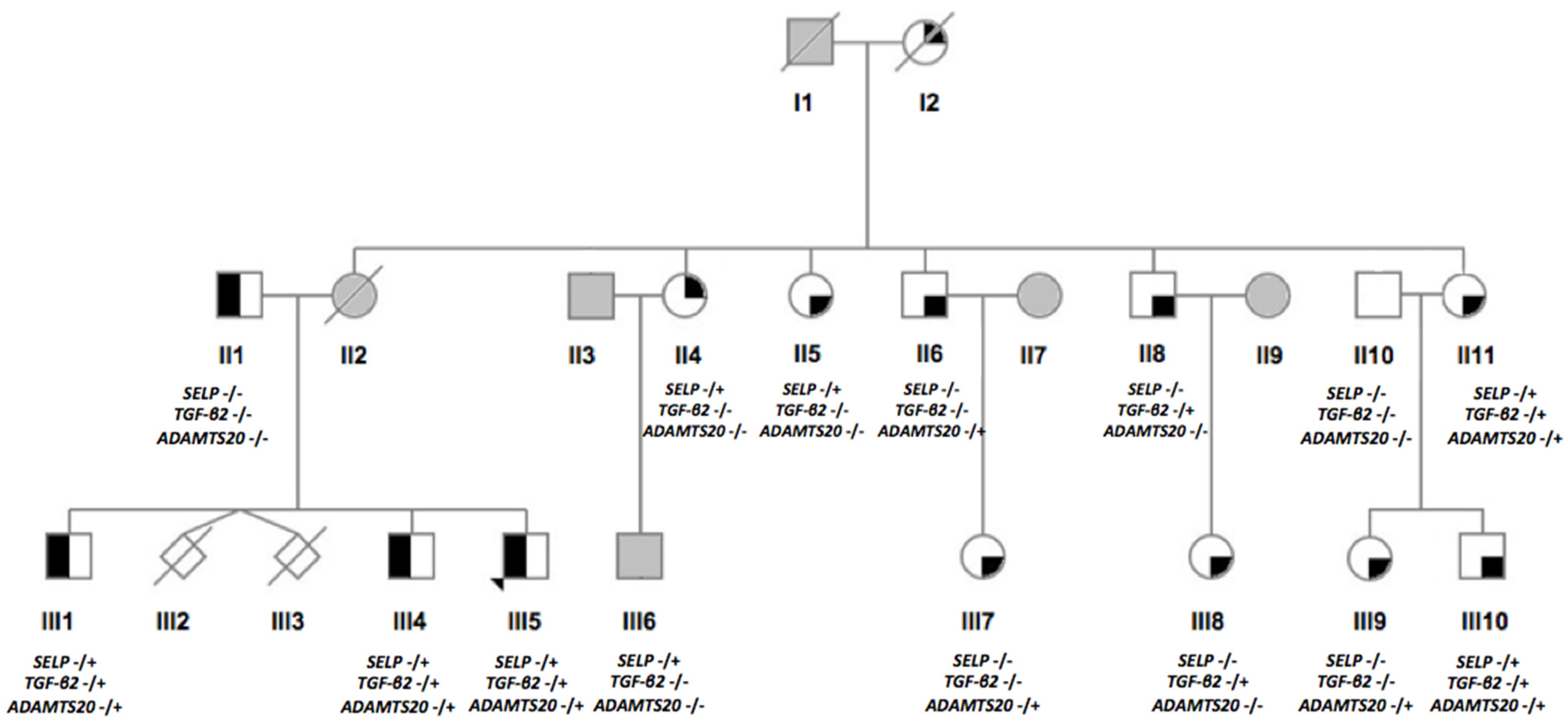
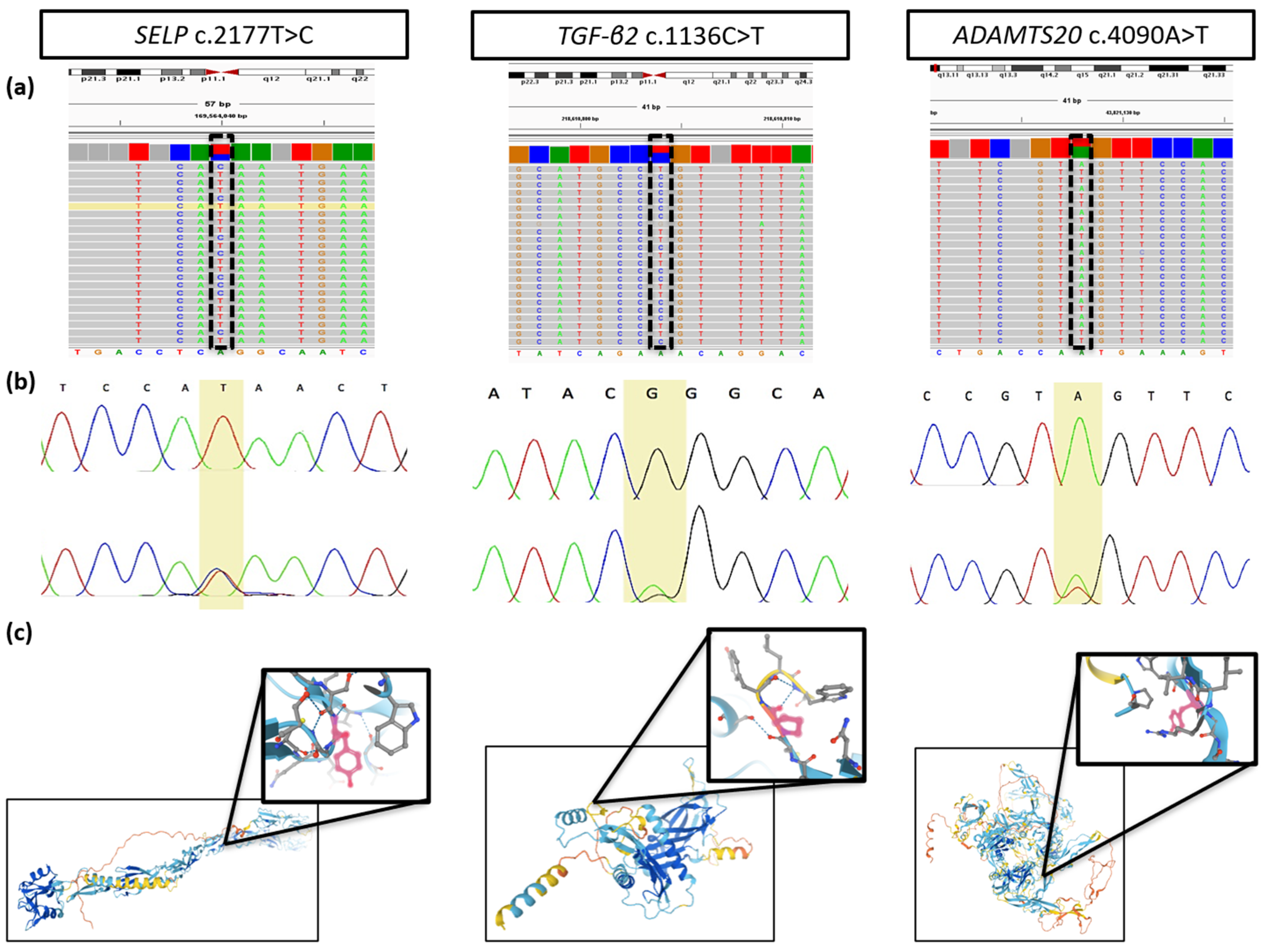
3.2. Description of the Identified Gene Variants
3.3. Genotyping Results of the Epidemiological Study
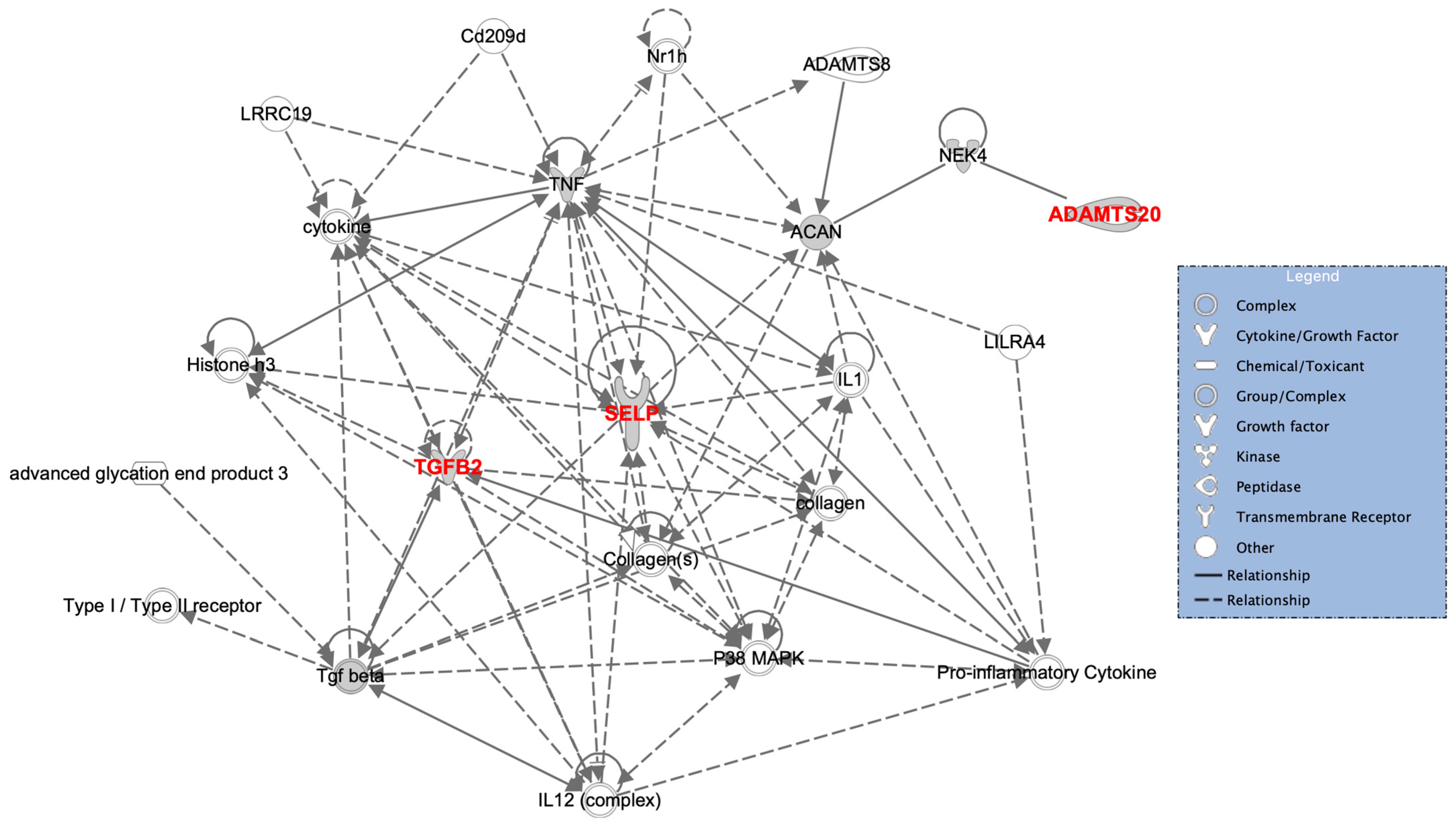
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ralston, S.H.; de Crombrugghe, B. Genetic regulation of bone mass and susceptibility to osteoporosis. Genes Dev. 2006, 20, 2492–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, E.L.; Brown, M.A. Clinical review 2: Genetic determinants of bone density and fracture risk--state of the art and future directions. J. Clin. Endocrinol. Metab. 2010, 95, 2576–2587. [Google Scholar] [CrossRef] [PubMed]
- Dimai, H.P. Use of dual-energy X-ray absorptiometry (DXA) for diagnosis and fracture risk assessment; WHO-criteria, T- and Z-score, and reference databases. Bone 2017, 104, 39–43. [Google Scholar] [CrossRef]
- Foessl, I.; Bassett, J.H.D.; Bjørnerem, Å.; Busse, B.; Calado, Â.; Chavassieux, P.; Christou, M.; Douni, E.; Fiedler, I.A.K.; Fonseca, J.E.; et al. Bone Phenotyping Approaches in Human, Mice and Zebrafish—Expert Overview of the EU Cost Action GEMSTONE (“GEnomics of MusculoSkeletal traits TranslatiOnal NEtwork”). Front. Endocrinol. 2021, 12, 720728. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Ferrari, S. IOF Compendium of Osteoporosis; Universitat de Geneve: Geneve Switzerland, 2019; pp. 1–90. [Google Scholar]
- Drake, M.T.; Clarke, B.L.; Lewiecki, E.M. The Pathophysiology and Treatment of Osteoporosis. Clin. Ther. 2015, 37, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Ongphiphadhanakul, B. Osteoporosis: The role of genetics and the environment. Forum Nutr. 2007, 60, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-de Sosa, N.; Athanasiadis, G.; Malouf, J.; Laiz, A.; Marin, A.; Herrera, S.; Farrerons, J.; Soria, J.M.; Casademont, J. Heritability of bone mineral density in a multivariate family-based study. Calcif. Tissue Int. 2014, 94, 590–596. [Google Scholar] [CrossRef]
- Yang, T.L.; Shen, H.; Liu, A.; Dong, S.S.; Zhang, L.; Deng, F.Y.; Zhao, Q.; Deng, H.W. A road map for understanding molecular and genetic determinants of osteoporosis. Nat. Rev. Endocrinol. 2020, 16, 91–103. [Google Scholar] [CrossRef]
- Koromani, F.; Trajanoska, K.; Rivadeneira, F.; Oei, L. Recent Advances in the Genetics of Fractures in Osteoporosis. Front. Endocrinol. 2019, 10, 337. [Google Scholar] [CrossRef] [Green Version]
- Formosa, M.M.; Bergen, D.J.M.; Gregson, C.L.; Maurizi, A.; Kämpe, A.; Garcia-Giralt, N.; Zhou, W.; Grinberg, D.; Ovejero Crespo, D.; Zillikens, M.C.; et al. A Roadmap to Gene Discoveries and Novel Therapies in Monogenic Low and High Bone Mass Disorders. Front. Endocrinol. 2021, 12, 709711. [Google Scholar] [CrossRef]
- Korvala, J.; Jüppner, H.; Mäkitie, O.; Sochett, E.; Schnabel, D.; Mora, S.; Bartels, C.F.; Warman, M.L.; Deraska, D.; Cole, W.G.; et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med. Genet. 2012, 13, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, C.M.; Chung, B.D.; Susic, M.; Prescott, T.; Semler, O.; Fiskerstrand, T.; D’Eufemia, P.; Castori, M.; Pekkinen, M.; Sochett, E.; et al. Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG). Eur J. Hum. Genet. 2011, 19, 875–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.L.; Deutsch, S.; Choudhury, U.; Chevalley, T.; Bonjour, J.P.; Dermitzakis, E.T.; Rizzoli, R.; Antonarakis, S.E. Polymorphisms in the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with variation in vertebral bone mass, vertebral bone size, and stature in whites. Am. J. Hum. Genet. 2004, 74, 866–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitjaroentham, A.; Hananantachai, H.; Phonrat, B.; Preutthipan, S.; Tungtrongchitr, R. Low density lipoprotein receptor-related protein 5 gene polymorphisms and osteoporosis in Thai menopausal women. J. Negat Results Biomed. 2016, 15, 16. [Google Scholar] [CrossRef] [Green Version]
- Sassi, R.; Sahli, H.; Souissi, C.; El Mahmoudi, H.; Zouari, B.; Ben Ammar ElGaaied, A.; Sellami, S.; Ferrari, S.L. Association of LRP5 genotypes with osteoporosis in Tunisian post-menopausal women. BMC Musculoskelet. Disord. 2014, 15, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.-Y.; Qiu, Y.; Mao, H.-J. Common Polymorphism in the LRP5 Gene May Increase the Risk of Bone Fracture and Osteoporosis. BioMed. Res. Int. 2014, 2014, 290531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Cai, Y.; Yao, Z.; Lin, J. Genetic analysis of the relationship between bone mineral density and low-density lipoprotein receptor-related protein 5 gene polymorphisms. PLoS ONE 2013, 8, e85052. [Google Scholar] [CrossRef]
- Laine, C.M.; Joeng, K.S.; Campeau, P.M.; Kiviranta, R.; Tarkkonen, K.; Grover, M.; Lu, J.T.; Pekkinen, M.; Wessman, M.; Heino, T.J.; et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013, 368, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, F.S.; Zillikens, M.C.; Micha, D.; Riessland, M.; Marcelis, C.L.; de Die-Smulders, C.E.; Milbradt, J.; Franken, A.A.; Harsevoort, A.J.; Lichtenbelt, K.D.; et al. PLS3 mutations in X-linked osteoporosis with fractures. N. Engl. J. Med. 2013, 369, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.; Rizzoli, R.; Bonjour, J.P. Heritable and nutritional influences on bone mineral mass. Aging 1998, 10, 205–213. [Google Scholar] [CrossRef]
- Michaëlsson, K.; Melhus, H.; Ferm, H.; Ahlbom, A.; Pedersen, N.L. Genetic liability to fractures in the elderly. Arch. Intern. Med. 2005, 165, 1825–1830. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Physical Status: The Use of and Interpretation of Anthropometry, Report of a WHO Expert Committee; World Health Organization (WHO): Geneve, Switzerland, 1995.
- World Health Organization (WHO). Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation; World Health Organization (WHO): Geneve, Switzerland, 2000.
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Spies, N.; Weng, Z.; Bishara, A.; McDaniel, J.; Catoe, D.; Zook, J.M.; Salit, M.; West, R.B.; Batzoglou, S.; Sidow, A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 2017, 14, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. In Current Protocols in Human Genetics; Wiley Online Library: Hoboken, NJ, USA, 2013; Chapter 7, Unit 7.20. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat 2016, 37, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat 2015, 36, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meynert, A.M.; Ansari, M.; FitzPatrick, D.R.; Taylor, M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinform. 2014, 15, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes Fajardo, K.V.; Adams, D.; Mason, C.E.; Sincan, M.; Tifft, C.; Toro, C.; Boerkoel, C.F.; Gahl, W.; Markello, T. Detecting false-positive signals in exome sequencing. Hum. Mutat 2012, 33, 609–613. [Google Scholar] [CrossRef] [Green Version]
- Shyr, C.; Tarailo-Graovac, M.; Gottlieb, M.; Lee, J.J.; van Karnebeek, C.; Wasserman, W.W. FLAGS, frequently mutated genes in public exomes. BMC Med. Genom. 2014, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Hayamizu, T.F.; Finger, J.H.; Bello, S.M.; McCright, I.J.; Xu, J.; Baldarelli, R.M.; Beal, J.S.; Campbell, J.; Corbani, L.E.; et al. The mouse Gene Expression Database (GXD): 2019 update. Nucleic Acids Res. 2019, 47, D774–D779. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Fuentes, V.; Cacheiro, P.; Meehan, T.F.; Aguilar-Pimentel, J.A.; Brown, S.D.M.; Flenniken, A.M.; Flicek, P.; Galli, A.; Mashhadi, H.H.; Hrabě de Angelis, M.; et al. The International Mouse Phenotyping Consortium (IMPC): A functional catalogue of the mammalian genome that informs conservation. Conserv. Genet. 2018, 19, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Kiel, D.P.; Kemp, J.P.; Rivadeneira, F.; Westendorf, J.J.; Karasik, D.; Duncan, E.; Imai, Y.; Müller, R.; Flannick, J.; Bonewald, L.; et al. The Musculoskeletal Knowledge Portal: Making Omics Data Useful to the Broader Scientific Community. J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef]
- Formosa, M.M.; Xuereb-Anastasi, A. Biochemical Predictors of Low Bone Mineral Density and Fracture Susceptibility in Maltese Postmenopausal Women. Calcif. Tissue Int. 2016, 98, 28–41. [Google Scholar] [CrossRef]
- Christiansen, B.A.; Harrison, S.L.; Fink, H.A.; Lane, N.E. Incident fracture is associated with a period of accelerated loss of hip BMD: The Study of Osteoporotic Fractures. Osteoporos. Int. 2018, 29, 2201–2209. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Chen, M.; Geng, J.G. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch. Immunol. Ther. Exp. 2006, 54, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, B.; Lee, J.; Mortensen, L.J.; Yu, H.; Wu, Z.L.; Lin, C.P.; Rossi, D.J.; Sackstein, R. Glycoengineering of E-Selectin Ligands by Intracellular versus Extracellular Fucosylation Differentially Affects Osteotropism of Human Mesenchymal Stem Cells. Stem Cells 2016, 34, 2501–2511. [Google Scholar] [CrossRef] [Green Version]
- Reid, K.B.; Day, A.J. Structure-function relationships of the complement components. Immunol. Today 1989, 10, 177–180. [Google Scholar] [CrossRef]
- Bullard, D.C.; Mobley, J.M.; Justen, J.M.; Sly, L.M.; Chosay, J.G.; Dunn, C.J.; Lindsey, J.R.; Beaudet, A.L.; Staite, N.D. Acceleration and increased severity of collagen-induced arthritis in P-selectin mutant mice. J. Immunol. 1999, 163, 2844–2849. [Google Scholar]
- Tu-Rapp, H.; Pu, L.; Marques, A.; Kulisch, C.; Yu, X.; Gierer, P.; Ibrahim, S.M.; Vollmar, B. Genetic control of leucocyte--endothelial cell interaction in collagen-induced arthritis. Ann. Rheum. Dis. 2010, 69, 606–610. [Google Scholar] [CrossRef]
- André, P. P-selectin in haemostasis. Br. J. Haematol. 2004, 126, 298–306. [Google Scholar] [CrossRef]
- Geng, J.G.; Chen, M.; Chou, K.C. P-selectin cell adhesion molecule in inflammation, thrombosis, cancer growth and metastasis. Curr. Med. Chem. 2004, 11, 2153–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, L.A.; Anderson, C.J.; Novelli, E.M. Targeting P-Selectin Adhesion Molecule in Molecular Imaging: P-Selectin Expression as a Valuable Imaging Biomarker of Inflammation in Cardiovascular Disease. J. Nucl. Med. 2019, 60, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Krauser, A.F.; Ponnarasu, S.; Schury, M.P. Holt Oram Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Alhamdi, S.; Lee, Y.C.; Chowdhury, S.; Byers, P.H.; Gottschalk, M.; Taft, R.J.; Joeng, K.S.; Lee, B.H.; Bird, L.M. Heterozygous WNT1 variant causing a variable bone phenotype. Am. J. Med. Genet. A 2018, 176, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Lobo, I. Same genetic mutation, different genetic disease phenotype. Nature 2008, 1, 64. [Google Scholar]
- Jamsheer, A.; Sowińska-Seidler, A.; Olech, E.M.; Socha, M.; Kozłowski, K.; Pyrkosz, A.; Trzeciak, T.; Materna-Kiryluk, A.; Latos-Bieleńska, A. Variable expressivity of the phenotype in two families with brachydactyly type E, craniofacial dysmorphism, short stature and delayed bone age caused by novel heterozygous mutations in the PTHLH gene. J. Hum. Genet. 2016, 61, 457–461. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.-L.; Chen, Z.-H.; Teng, Y.-Y.; Liu, S.-Y.; Jia, Y.; Zhang, K.-W.; Sun, Z.-L.; Wu, J.-J.; Yuan, Z.-D.; Feng, Y.; et al. The Smad Dependent TGF-β and BMP Signaling Pathway in Bone Remodeling and Therapies. Front. Mol. Biosci. 2021, 8, 389. [Google Scholar] [CrossRef]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int J. Mol. Sci. 2020, 21, 7597. [Google Scholar] [CrossRef]
- Finnson, K.W.; Chi, Y.; Bou-Gharios, G.; Leask, A.; Philip, A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci 2012, 4, 251–268. [Google Scholar] [CrossRef]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.W.D.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, H.; Lutz, B.; Reicherter, K.; Lühl, S.; Taurman, R.; Gabriel, H.; Brenner, R.E.; Borck, G. 4.7 Mb deletion encompassing TGFB2 associated with features of Loeys-Dietz syndrome and osteoporosis in adulthood. Am. J. Med. Genet. A 2017, 173, 2289–2292. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Freedman, M.L.; Reich, D.; Penney, K.L.; McDonald, G.J.; Mignault, A.A.; Patterson, N.; Gabriel, S.B.; Topol, E.J.; Smoller, J.W.; Pato, C.N.; et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004, 36, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Capelli, C.; Redhead, N.; Romano, V.; Calì, F.; Lefranc, G.; Delague, V.; Megarbane, A.; Felice, A.E.; Pascali, V.L.; Neophytou, P.I.; et al. Population structure in the Mediterranean basin: A Y chromosome perspective. Ann. Hum. Genet. 2006, 70, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.T.; Hayes, M.G. Testing the null hypothesis: Comments on ‘Culture-gene coevolution of individualism-collectivism and the serotonin transporter gene’. Proc. Biol. Sci. 2011, 278, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Koziell, A.; Grech, V.; Hussain, S.; Lee, G.; Lenkkeri, U.; Tryggvason, K.; Scambler, P. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum. Mol. Genet. 2002, 11, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, R.; Scerri, C.A.; Montalto, S.A.; Parascandolo, R.; Neville, B.G.; Felice, A.E. Molecular genetics of tetrahydrobiopterin (BH4) deficiency in the Maltese population. Mol. Genet. Metab. 2007, 90, 277–283. [Google Scholar] [CrossRef]
- Farrugia Wismayer, M.; Farrugia Wismayer, A.; Pace, A.; Vassallo, N.; Cauchi, R.J. SOD1 D91A variant in the southernmost tip of Europe: A heterozygous ALS patient resident on the island of Gozo. Eur. J. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Vidal, C.; Cachia, A.; Xuereb-Anastasi, A. Effects of a synonymous variant in exon 9 of the CD44 gene on pre-mRNA splicing in a family with osteoporosis. Bone 2009, 45, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [Green Version]
- Hubmacher, D.; Apte, S.S. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015, 47, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Mead, T.J.; Apte, S.S. ADAMTS proteins in human disorders. Matrix Biol. 2018, 71–72, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.P.; He, Y.Z. Bone growth during daily or intermittent calcitriol treatment during renal failure with advanced secondary hyperparathyroidism. Kidney Int. 2007, 72, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Roughley, P.J.; Mort, J.S. The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 2014, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.; Foernzler, D.; Loftus, S.K.; Liu, S.; McPherson, J.D.; Jungers, K.A.; Apte, S.S.; Pavan, W.J.; Beier, D.R. A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development 2003, 130, 4665–4672. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, H.; Nelson, C.M.; Somerville, R.P.; Mielke, K.; Dixon, L.J.; Powell, K.; Apte, S.S. Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development 2010, 137, 4029–4038. [Google Scholar] [CrossRef] [Green Version]
- Holdener, B.C.; Percival, C.J.; Grady, R.C.; Cameron, D.C.; Berardinelli, S.J.; Zhang, A.; Neupane, S.; Takeuchi, M.; Jimenez-Vega, J.C.; Uddin, S.M.Z.; et al. ADAMTS9 and ADAMTS20 are differentially affected by loss of B3GLCT in mouse model of Peters plus syndrome. Hum. Mol. Genet 2019, 28, 4053–4066. [Google Scholar] [CrossRef]
- Adams, J.C. Thrombospondins: Multifunctional regulators of cell interactions. Annu. Rev. Cell Dev. Biol. 2001, 17, 25–51. [Google Scholar] [CrossRef]
- Zhang, L. Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog. Mol. Biol. Transl. Sci. 2010, 93, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [Green Version]
- Maroulakou, I.G.; Bowe, D.B. Expression and function of Ets transcription factors in mammalian development: A regulatory network. Oncogene 2000, 19, 6432–6442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raouf, A.; Seth, A. Ets transcription factors and targets in osteogenesis. Oncogene 2000, 19, 6455–6463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Rifkin, S.A.; Andersen, E.; van Oudenaarden, A. Variability in gene expression underlies incomplete penetrance. Nature 2010, 463, 913–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, I. Phenotype Variability: Penetrance and Expressivity. Nat. Educ. 2008, 1, 137. [Google Scholar]
- Napoli, N.; Faccio, R.; Shrestha, V.; Bucchieri, S.; Rini, G.B.; Armamento-Villareal, R. Estrogen metabolism modulates bone density in men. Calcif. Tissue Int. 2007, 80, 227–232. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 2013, 161, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ponthieux, A.; Herbeth, B.; Droesch, S.; Haddy, N.; Lambert, D.; Visvikis, S. Biological determinants of serum ICAM-1, E-selectin, P-selectin and L-selectin levels in healthy subjects: The Stanislas study. Atherosclerosis 2004, 172, 299–308. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; Rudi, T.; von Baehr, V. Osteoimmunology of tumor necrosis factor-alpha, IL-6, and RANTES/CCL5: A review of known and poorly understood inflammatory patterns in osteonecrosis. Clin. Cosmet Investig Dent. 2018, 10, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, M.B.; Shim, J.H. Osteoimmunology: A brief introduction. Immune Netw. 2013, 13, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-α contributes to postmenopausal osteoporosis by synergistically promoting RANKL-induced osteoclast formation. Biomed. Pharm. 2018, 102, 369–374. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age (Years) | BMI (kg/m2) | LS T-Score | LS Z-Score | FN T-Score | FN Z-Score | TH T-Score | TH Z-Score | Fracture Sustained | WGS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II1 | M | 74 | 27.9 | −2.6 | −1.6 | −1.7 | −0.3 | −0.7 | 0.1 | - | ✓ |
| II4 | F | 72 | - | - | - | - | - | - | - | Wrist at 70 years | ✓ |
| II5 | F | 71 | 30 | −1.3 | 1.0 | −0.6 | 1.3 | 0.1 | 1.6 | - | |
| II7 | M | 69 | 24.8 | −1.6 | −0.7 | −1.7 | −0.5 | −1.4 | −0.8 | - | ✓ |
| II8 | M | 66 | 27.9 | −1.7 | −0.8 | −1.8 | −0.7 | −0.9 | −0.3 | - | ✓ |
| II10 | M | 69 | 36.9 | 1.8 | 2.7 | −0.4 | 0.7 | 0.1 | 0.7 | - | ✓ |
| II11 | F | 63 | 35.6 | −1.4 | 0.3 | −0.4 | 1.1 | −0.2 | 0.9 | - | |
| III1 | M | 47 | 21.0 | −2.2 | −1.8 | −2.9 | −2.2 | −1.9 | −1.5 | - | ✓ |
| III4 | M | 36 | 22.5 | −2.5 | −2.4 | −1.5 | −0.8 | −1.2 | −1.0 | - | ✓ |
| III5 | M | 32 | 22.5 | −3.7 | −3.6 | −2.6 | −2.3 | −1.7 | −1.5 | - | ✓ |
| III6 | M | 52 | - | - | - | - | - | - | - | - | ✓ |
| III7 | F | 42 | 24.7 | −1.6 | −1.2 | −1.3 | −1.0 | −1.3 | −1.0 | - | ✓ |
| III8 | F | 28 | 18.3 | −1.3 | −1.3 | −1.5 | −1.4 | −1.4 | −1.4 | - | ✓ |
| III9 | F | 43 | 27.9 | −0.8 | −0.4 | −1.7 | −0.3 | −0.7 | 0.1 | - | ✓ |
| III10 | M | 39 | 54.8 | −1.3 | −1.3 | 0.8 | 1.2 | 0.0 | 0.2 | - |
| Filtering Step | Single Nucleotide Variants (SNVs) | Insertions & Deletions (InDels) |
|---|---|---|
| Total number of variants | 42,854 | 1957 |
| Zygosity filtering following a dominant inheritance pattern for affected relatives (III1, III4, III5); removal of all variants fitting a dominant or recessive inheritance pattern in the unaffected control (II10) | 1902 | 55 |
| Removal of benign variants including synonymous, deep intronic variants | 936 | 18 |
| Retaining of gene variants with an observed alternative allele frequency of ≤1% in population-based allele frequency databases | 34 | 18 |
| Retaining of missense variants predicted to be deleterious by most in silico prediction tools | 33 | 18 |
| Removal of ‘frequent hitters’ | 32 | 18 |
| Removal of genes with no association to bone or fractures in literature | 3 | 0 |
| Gene | Variant (Rs Number) | AAF (GnomAD) | Chromosome: Position 1 | Coverage | Amino Acid Change | CADD Score | Polyphen-2 | SIFT | Mutation Taster | Mutation Assessor | Vest3 | Clinvar | ACMG Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SELP | c.2177T>C (rs754086574) | C: 0.0000756 | 1: 169564040 | 49 | p.Tyr726Cys | 23.5 | D | D | D | D | D | NR | 4 2 |
| TGF-β2 | c.1136C>T (rs773943154) | T: 0.0000159 | 1: 218610804 | 47 | p.Pro379Leu | 25.0 | D | D | D | D | D | US | 4 |
| ADAMTS20 | c.4090A>T (rs138035327) | T: 0.0008091 | 12: 43821128 | 46 | p.Tyr1364Asn | 24.3 | D | B | D | D | D | NR | 4 |
| Variant | Genotype | AAF | HWE 1 | ||
|---|---|---|---|---|---|
| Homozygous Reference n (%) | Heterozygous n (%) | Homozygous Alternative n (%) | |||
| SELP c.2177T>C Controls Normal BMD Osteopenic BMD 2 Osteoporotic BMD 3 All-type Fractures Whole collection | TT: 226 (98.7) TT: 259 (97.4) TT: 280 (99.3) TT: 263 (98.1) TT: 1028 (98.4) | TC: 3 (1.3) TC: 3 (1.1) TC: 1 (0.4) TC: 4 (1.5) TC: 11 (1.1) | CC: 0 CC: 4 (1.5) CC: 1 (0.4) CC: 1 (0.4) CC: 6 (0.6) | C: 0.007 C: 0.021 C: 0.005 C: 0.011 C: 0.011 | p = 1.00 |
| TGF-β2 c.1136C>T Controls Normal BMD Osteopenic BMD 2 Osteoporotic BMD 3 All-type Fractures Whole collection | CC: 228 (99.6) CC: 265 (99.6) CC: 282 (100) CC: 265 (98.9) CC: 1040 (99.5) | CT: 1 (0.4) CT: 1 (0.4) CT: 0 (0.0) CT: 3 (1.1) CT: 5 (0.5) | TT: 0 TT: 0 TT: 0 TT: 0 TT: 0 | T: 0.002 T: 0.002 - T: 0.006 T: 0.002 | p = 1.00 |
| ADAMTS20 c.4090A>T Controls Normal BMD Osteopenic BMD 2 Osteoporotic BMD 3 All-type Fractures Whole collection | AA: 205 (94.0) AA: 247 (95.7) AA: 258 (93.5) AA: 245 (94.2) AA: 955 (94.4) | AT: 13 (6.0) AT: 11 (4.3) AT: 18 (6.5) AT: 15 (5.8) AT: 57 (5.6) | TT: 0 TT: 0 TT: 0 TT: 0 TT: 0 | T: 0.030 T: 0.021 T: 0.033 T: 0.029 T: 0.028 | p = 1.00 |
| Phenotype (Units) | Normal BMD 1 | Osteopenia & Osteoporosis 2 | All-Type Fractures | Whole Collection | ||||
|---|---|---|---|---|---|---|---|---|
| SELP c.2177T>C | TT (n = 226) | TC/CC (n= 3) | TT (n= 539) | TC/CC (n= 9) | TT (n= 263) | TC/CC (n= 5) | TT (n= 1028) | TC/CC (n= 17) |
| LS BMD (g/cm2) | 1.13 (1.05–1.23) | 1.36 (1.15–1.46) | 0.92 (0.84–0.99) | 0.95 (0.86–0.99) | 0.91 (0.82–1.01) | 0.91 (0.85–1.07) | 0.96 (0.86–1.07) | 0.99 (0.89–1.07) |
| FN BMD (g/cm2) | 0.89 (0.85–0.97) | 0.97 (0.89–0.99) | 0.74 (0.67–0.80) | 0.76 (0.71–0.82) | 0.69 (0.63–0.77) | 0.67 (0.58–0.70) | 0.76 (0.68–0.86) | 0.75 (0.70–0.86) |
| TH BMD (g/cm2) | 1.00 (0.91–1.06) | 0.98 (0.95–1.06) | 0.80 (0.73–0.88) | 0.84 (0.77–0.89) | 0.76 (0.70–0.85) | 0.64 (0.59–0.81) | 0.83 (0.74–0.92) | 0.85 (0.75–0.93) |
| Calcium (mmol/L) 3 | 2.40 (2.32–2.46) | 2.37 (2.35–2.44) | 2.40 (2.34–2.48) | 2.39 (2.38–2.53) | 2.34 (2.26–2.43) | 2.25 (2.14–2.41) | 2.39 (2.31–2.46) | 2.39 (2.33–2.44) |
| ALP (U/L) 4 | 165 (132–197) | 243 (128–288) | 157 (126–185) | 192 (163–218) | 154 (120–183) | 231 (94–243) | 157 (126–186) | 214 (153–234) |
| Albumin (g/L) 5 | 44.0 (42.2–45.9) | 47.1 (40.9–47.7) | 43.8 (41.9–45.8) | 43.5 (41.6–46.9) | 42.2 (39.9–44.6) | 43.4 (38.6–44.7) | 43.5 (41.6–45.6) | 43.4 (41.3–46.6) |
| TGF-β2 c.1136C>T | CC (n= 228) | CT (n= 1) | CC (n= 547) | CT (n= 1) | CC (n= 265) | CT (n= 3) | CC (n= 1040) | CT (n= 5) |
| LS BMD (g/cm2) | 1.13 (1.05–1.23) | 1.16 | 0.92 (0.84–0.99) | 0.87 | 0.91 (0.82–1.01) | 0.91 (0.71–0.96) | 0.96 (0.86–1.07) | 0.91 (0.79–1.04) |
| FN BMD (g/cm2) | 0.89 (0.85–0.97) | 1.00 | 0.73 (0.67–0.80) | 0.80 | 0.69 (0.63–0.77) | 0.63 (0.54–0.71) | 0.76 (0.68–0.86) | 0.71 (0.58–0.90) |
| TH BMD (g/cm2) | 0.99 (0.91–1.06) | 1.19 | 0.80 (0.74–0.88) | 0.82 | 0.76 (0.70–0.85) | 0.56 (0.56–0.71) | 0.83 (0.74–0.92) | 0.71 (0.56–1.00) |
| Calcium (mmol/L) | 2.40 (2.32–2.46) | 2.42 | 2.40 (2.34–2.48) | 2.55 | 2.34 (2.26–2.43) | 2.36 (2.15–2.43) | 2.39 (2.31–2.46) | 2.42 (2.26–2.50) |
| ALP (U/L) | 165 (131–198) | 164 | 158 (126–185) | 140 | 154 (119–184) | 170 (104–246) | 158 (127–187) | 164 (122–208) |
| Albumin (g/L) | 44.0 (42.2–45.9) | 46.2 | 43.8 (41.9–45.8) | 45.4 | 42.2 (39.9–44.6) | 42.6 (40.4–45.2) | 43.5 (41.6–45.6) | 45.2 (41.5–45.8) |
| ADAMTS20 c.4090A>T | AA (n= 205) | AT (n= 13) | AA (n= 505) | AT (n= 29) | AA (n= 245) | AT (n= 15) | AA (n= 955) | AT (n= 57) |
| LS BMD (g/cm2) | 1.13 (1.06–1.23) | 1.05 (1.04–1.18) | 0.92 (0.84–0.99) | 0.88 (0.83–1.02) | 0.91 (0.82–1.00) | 0.87 (0.74–0.99) | 0.96 (0.86–1.07) | 0.97 (0.84–1.05) |
| FN BMD (g/cm2) | 0.89 (0.85–0.97) | 0.88 (0.82–0.94) | 0.74 (0.67–0.79) | 0.74 (0.66–0.79) | 0.69 (0.63–0.76) | 0.73 (0.66–0.78) | 0.76 (0.68–0.85) | 0.76 (0.68–0.82) |
| TH BMD (g/cm2) | 1.00 (0.91–1.06) | 0.91 (0.84–0.98) | 0.81 (0.74–0.88) | 0.78 (0.73–0.87) | 0.75 (0.69–0.85) | 0.81 (0.73–0.83) | 0.83 (0.74–0.92) | 0.82 (0.75–0.90) |
| Calcium (mmol/L) | 2.40 (2.32–2.47) | 2.37 (2.29–2.41) | 2.41 (2.34–2.48) | 2.38 (2.34–2.43) | 2.35 (2.26–2.43) | 2.33 (2.27–2.37) | 2.39 (2.31–2.46) | 2.37 (2.33–2.41) |
| ALP (U/L) | 165 (132–198) | 167 (139-196) | 159 (128–188) | 152 (117–166) | 154 (120–184) | 155 (108-209) | 158 (128–187) | 155 (120–182) |
| Albumin (g/L) | 44.0 (42.2–46.0) | 43.8 (42.5–45.2) | 43.7 (41.8–45.7) | 44.6 (42.4–47.4) | 42.3 (40.0–44.7) | 41.9 (40.3–44.5) | 43.5 (41.6–45.6) | 43.8 (41.9–46.3) |
| Genotype | Normal BMD n (%) | LS BMD n (%) 1 | Crude OR (95% CI), p | Adjusted OR (95% CI), p 2 |
| SELP c.2177T>C TT | 331 (98.8) | 4 (1.2) | 1.0 | 1.0 |
| TC/CC | 434 (98.2) | 8 (1.8) | 1.53 (0.46–5.11), 0.494 | 1.55 (0.46–5.20), 0.482 |
| ADAMTS20 c.4090A>T AA | 301 (93.8) | 409 (94.9) | 1.0 | 1.0 |
| AT | 20 (6.2) | 22 (5.1) | 0.81 (0.43–1.51), 0.507 | 0.77 (0.41–1.44), 0.415 |
| Genotype | Normal BMD n (%) | FN BMD n (%) 3 | Crude OR (95% CI), p | Adjusted OR (95% CI), p |
| SELP c.2177T>C | ||||
| TT | 318 (98.1) | 6 (1.9) | 1.0 | 1.0 |
| TC/CC | 447 (98.7) | 6 (1.3) | 0.71 (0.23–2.23), 0.558 | 0.72 (0.23–2.27), 0.570 |
| ADAMTS20 c.4090A>T AA | 293 (94.8 | 16 (5.2) | 1.0 | 1.0 |
| AT | 417 (94.1) | 26 (5.9) | 1.14 (0.60–2.17), 0.685 | 1.06 (0.55–2.03), 0.854 |
| Genotype | Normal BMD n (%) | TH BMD n (%) 4 | Crude OR (95% CI), p | Adjusted OR (95% CI), p |
| SELP c.2177T>C TT | 459 (98.3) | 8 (1.7) | 1.0 | 1.0 |
| TC/CC | 306 (98.7) | 4 (1.3) | 0.75 (0.22–2.51), 0.641 | 0.76 (0.22–2.57), 0.656 |
| ADAMTS20 c.4090A>T AA | 426 (94.7) | 24 (5.3) | 1.0 | 1.0 |
| AT | 284 (94.0) | 18 (6.0) | 1.13 (0.60–2.11), 0.714 | 1.04 (0.55–1.97), 0.909 |
| Genotype | Controls n (%) 5 | All-type Fractures n (%) | Crude OR (95% CI), p | Adjusted OR (95% CI), p 6 |
| SELP c.2177T>C TT | 765 (98.5) | 12 (1.5) | 1.0 | 1.0 |
| TC/CC | 263 (98.1) | 5 (1.9) | 1.21 (0.43–3.47), 0.720 | 1.16 (0.37–3.58), 0.800 |
| ADAMTS20 c.4090A>T AA AT | 710 (94.4) 245 (94.2) | 42 (5.6) 15 (5.8) | 1.0 1.04 (0.56–1.90), 0.912 | 1.0 1.02 (0.54–1.94), 0.947 |
| Genotype | Controls n (%) 5 | Wrist fractures n (%) 7 | Crude OR (95% CI), p | Adjusted OR (95% CI), p 6 |
| ADAMTS20 c.4090A>T AA AT | 710 (94.4) 107 (93.9) | 42 (5.6) 7 (6.1) | 1.0 1.11 (0.48–2.52), 0.811 | 1.0 1.12 (0.48–2.58), 0.799 |
| Calcium Tertile Intervals | ADAMTS20 c.4090A>T | Crude OR (95% CI), p | Adjusted OR (95% CI), p 1 | |
|---|---|---|---|---|
| AA | AT | |||
| Highest | 342 (97.2) | 10 (2.8) | 1.0 | |
| Intermediate | 285 (91.9) | 25 (8.1) | 3.00 (1.72–6.35), 0.004 | 2.99 (1.41–6.34), 0.004 |
| Lowest | 328 (93.7) | 22 (6.3) | 2.29 (1.07–4.92), <0.001 | 2.33 (1.08–5.03), 0.032 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilia, C.; Friggieri, D.; Vassallo, J.; Xuereb-Anastasi, A.; Formosa, M.M. Whole Genome Sequencing Unravels New Genetic Determinants of Early-Onset Familial Osteoporosis and Low BMD in Malta. Genes 2022, 13, 204. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020204
Cilia C, Friggieri D, Vassallo J, Xuereb-Anastasi A, Formosa MM. Whole Genome Sequencing Unravels New Genetic Determinants of Early-Onset Familial Osteoporosis and Low BMD in Malta. Genes. 2022; 13(2):204. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020204
Chicago/Turabian StyleCilia, Chanelle, Donald Friggieri, Josanne Vassallo, Angela Xuereb-Anastasi, and Melissa Marie Formosa. 2022. "Whole Genome Sequencing Unravels New Genetic Determinants of Early-Onset Familial Osteoporosis and Low BMD in Malta" Genes 13, no. 2: 204. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020204