
Possible Catch-Up Developmental Trajectories for Children with Mild Developmental Delay Caused by NAA15 Pathogenic Variants


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Compliance
2.2. Subjects
2.3. Neurodevelopmental Evaluation
2.4. Whole-Exome Sequencing (WES) and Variant Analysis
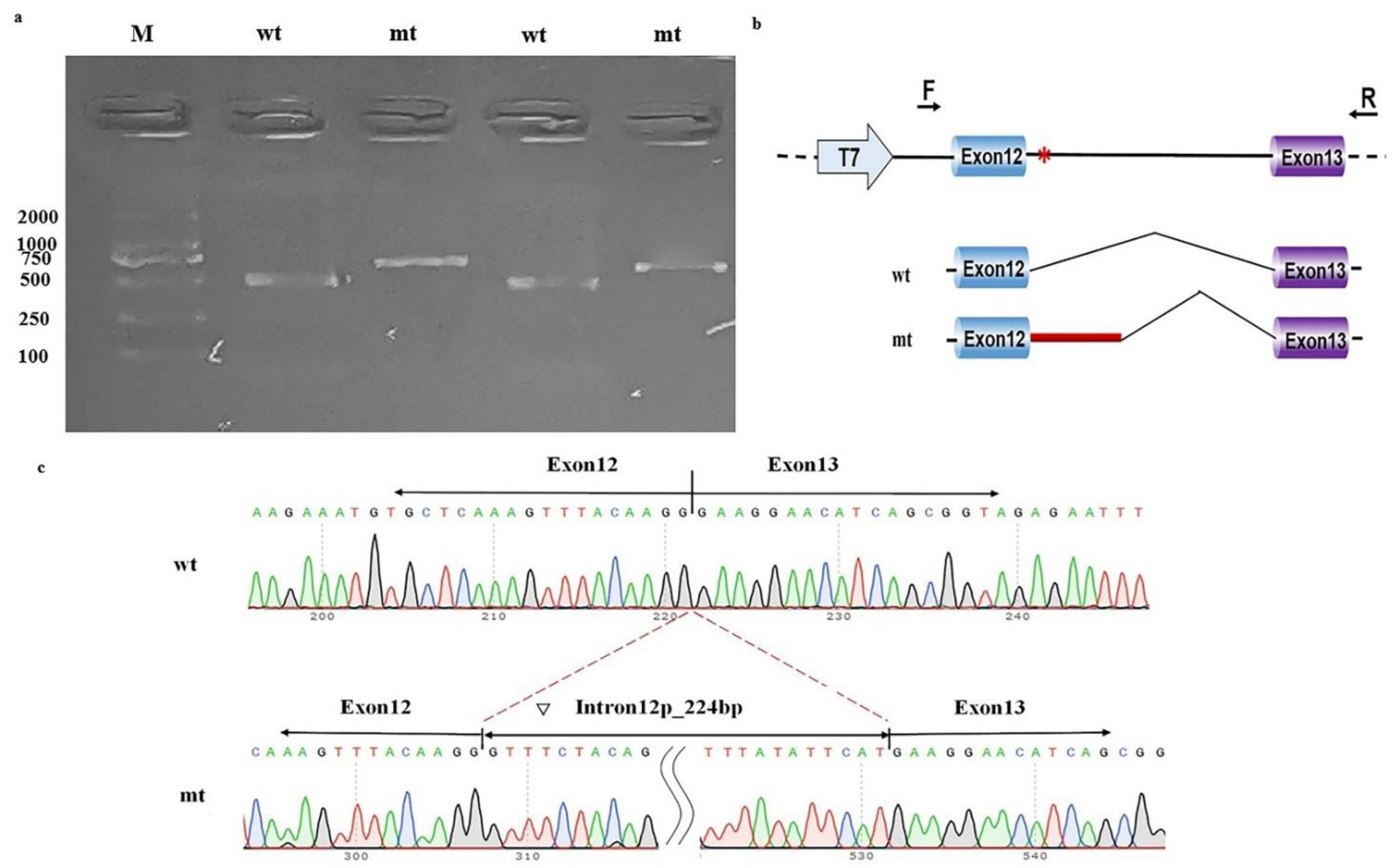
2.5. Functional Study of the Putative Splicing Variant in NAA15
3. Results
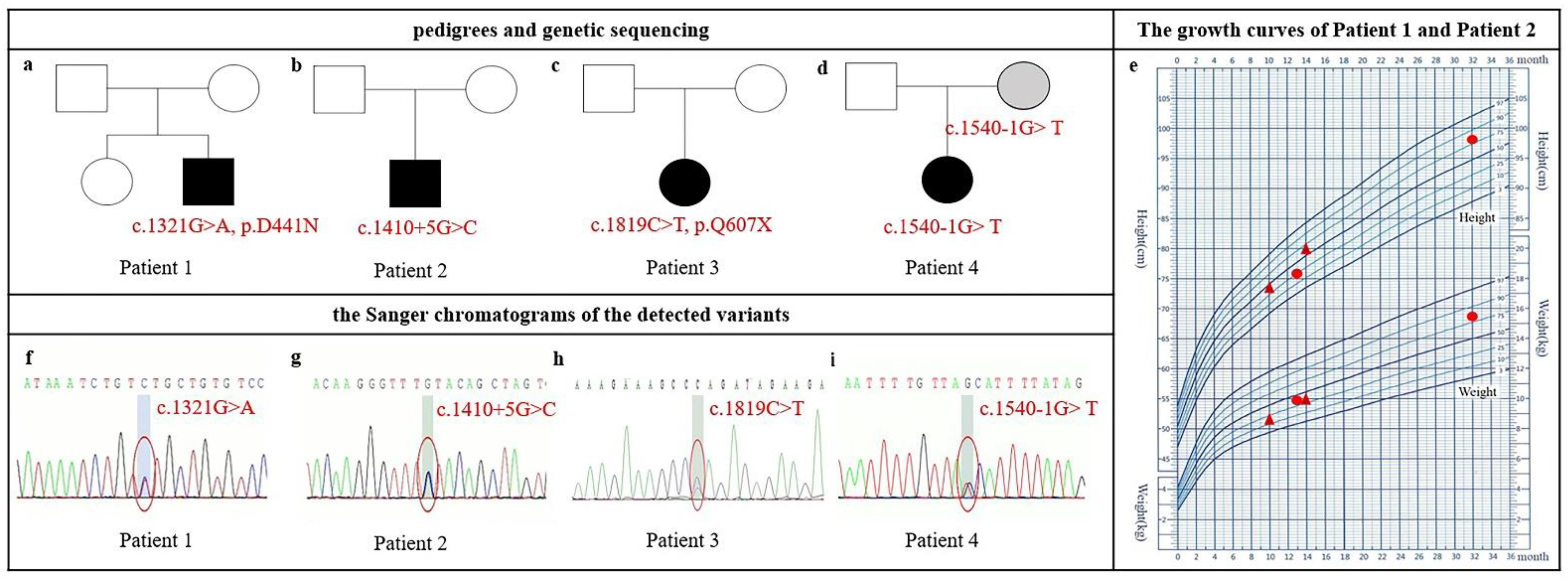
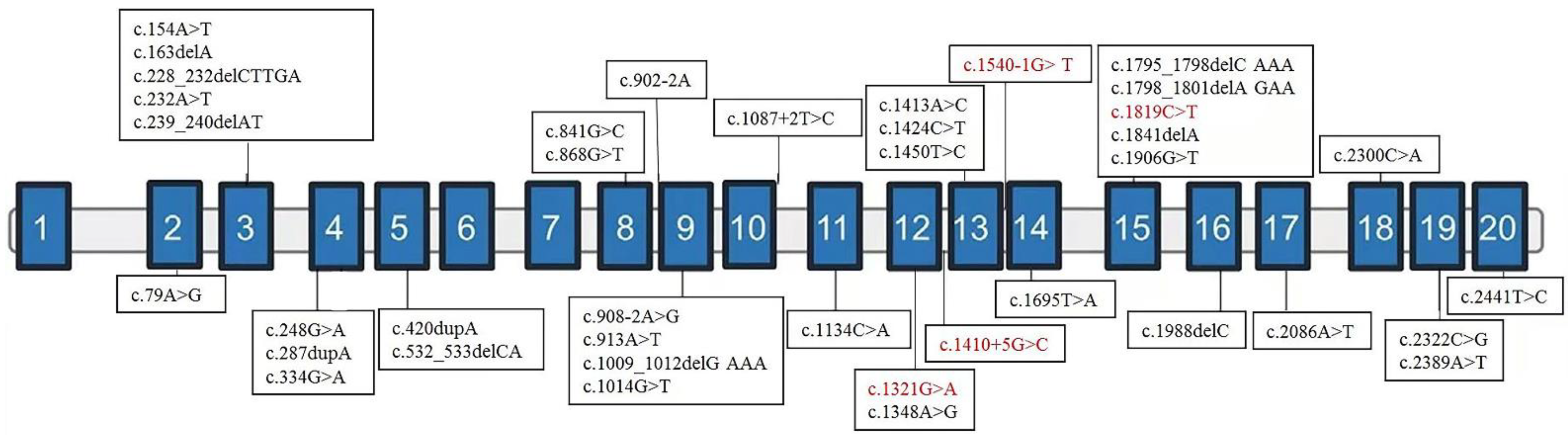
3.1. Clinical Features and Variant Spectrum of NAA15
3.2. Minigene Splicing Assay for the Noncanonical Splice Variant (c.1410+5G>C) in NAA15
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Moeschler, J.B.; Shevell, M.; Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [Green Version]
- Association, A.P. Intellectual disabilities. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Van Damme, P.; Evjenth, R.; Foyn, H.; Demeyer, K.; De Bock, P.J.; Lillehaug, J.R.; Vandekerckhove, J.; Arnesen, T.; Gevaert, K. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(alpha)-acetyltransferases and point to hNaa10p as the post-translational actin N(alpha)-acetyltransferase. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; McTiernan, N.; Wei, X.; Arnesen, T.; Marmorstein, R. Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat. Commun. 2020, 11, 818. [Google Scholar] [CrossRef] [Green Version]
- Varland, S.; Arnesen, T. Investigating the functionality of a ribosome-binding mutant of NAA15 using Saccharomyces cerevisiae. BMC Res Notes 2018, 11, 404. [Google Scholar] [CrossRef] [Green Version]
- Fluge, O.; Bruland, O.; Akslen, L.A.; Varhaug, J.E.; Lillehaug, J.R. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene 2002, 21, 5056–5068. [Google Scholar] [CrossRef] [Green Version]
- Arnesen, T.; Anderson, D.; Baldersheim, C.; Lanotte, M.; Varhaug, J.E.; Lillehaug, J.R. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 2005, 386, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. de novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Zhao, J.J.; Halvardson, J.; Zander, C.S.; Zaghlool, A.; Georgii-Hemming, P.; Mansson, E.; Brandberg, G.; Savmarker, H.E.; Frykholm, C.; Kuchinskaya, E.; et al. Exome sequencing reveals NAA15 and PUF60 as candidate genes associated with intellectual disability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Dharmadhikari, A.V.; Varland, S.; Ma, N.; Domingo, D.; Kleyner, R.; Rope, A.F.; Yoon, M.; Stray-Pedersen, A.; Posey, J.E.; et al. Truncating Variants in NAA15 Are Associated with Variable Levels of Intellectual Disability, Autism Spectrum Disorder, and Congenital Anomalies. Am. J. Hum. Genet. 2018, 102, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.H.; Feng, J.Y.; Wang, B.; Zhang, Y.; Wang, C.X.; Jia, F.Y. Comparison Of The Children Neuropsychological And Behavior Scale And The Griffiths Mental Development Scales When Assessing The Development Of Children With Autism. Psychol. Res. Behav. Manag. 2019, 12, 973–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, E.T.; Bricker, D.D.; Potter, L. Ages & Stages Questionnaires: User’s Guide, 3rd ed.; Paul H. Brookes: Baltimore, MD, USA, 2009. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, X.; Liu, X. In Silico Prediction of Deleteriousness for Nonsynonymous and Splice-Altering Single Nucleotide Variants in the Human Genome. Methods Mol. Biol. 2017, 1498, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Gottlieb, L.; Marchi, E.; Kleyner, R.; Bhardwaj, P.; Rope, A.F.; Rosenheck, S.; Moutton, S.; Philippe, C.; Eyaid, W.; et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 2019, 28, 2900–2919. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Berger, J.H.; Deardorff, M.; Izumi, K.; Lin, K.Y.; Medne, L.; Ahrens-Nicklas, R.C. Variants in NAA15 cause pediatric hypertrophic cardiomyopathy. Am. J. Med. Genet. Part A 2020, 185, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, N.; Patel, R.G.; Corriveau, R.A. N-methyl-D-aspartate receptors regulate a group of transiently expressed genes in the developing brain. J. Biol. Chem. 2001, 276, 14257–14263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzer, K.; Cogne, B.; Hague, J.; Marcelis, C.L.; Mitter, D.; Oberndorff, K.; Park, S.M.; Ploos van Amstel, H.K.; Simonic, I.; van der Smagt, J.J.; et al. Haploinsufficiency of CUX1 Causes Nonsyndromic Global Developmental Delay With Possible Catch-up Development. Ann. Neurol. 2018, 84, 200–207. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Wai, H.A.; Lord, J.; Lyon, M.; Gunning, A.; Kelly, H.; Cibin, P.; Seaby, E.G.; Spiers-Fitzgerald, K.; Lye, J.; Ellard, S.; et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet. Med. 2020, 22, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, J.; Gallone, G.; Short, P.J.; McRae, J.F.; Ironfield, H.; Wynn, E.H.; Gerety, S.S.; He, L.; Kerr, B.; Johnson, D.S.; et al. Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019, 29, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truty, R.; Ouyang, K.; Rojahn, S.; Garcia, S.; Colavin, A.; Hamlington, B.; Freivogel, M.; Nussbaum, R.L.; Nykamp, K.; Aradhya, S. Spectrum of splicing variants in disease genes and the ability of RNA analysis to reduce uncertainty in clinical interpretation. Am. J. Hum. Genet. 2021, 108, 696–708. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Literature | Total | Percent | |
|---|---|---|---|---|---|---|---|
| Age (months) | 13 | 10 | 17 | 24 | |||
| Nucleotide change * | c.1321G>A | c.1410+5G>C | c.1819C>T | c.1540-1G>T | |||
| Genomic location | Chr4:140280960 | Chr4:140281054 | Chr4:140291430 | Chr4:140282877 | |||
| Amino acid change | p. D441N | − | p. Q607X | − | |||
| Inheritance | de novo | de novo | de novo | maternal | |||
| ACMG classification | LP | LP | P | LP | |||
| Developmental delay (DD) | + | + | + | + | 10/40 | 13/43 | 23.3 |
| Gross motor delay | + | + | + | + | |||
| Fine motor delay | + | − | + | − | |||
| Language delay | + | +/− | + | + | |||
| Personal–social behavior delay | + | + | + | + | |||
| Adaptive behavior delay | + | − | + | + | |||
| Mild/moderate DD | + | + | + | + | 22/42 | 26/46 | 56.5 |
| ADHD or behavioral issues | − | − | + | + | 35/41 | 37/45 | 82.2 |
| Seizures | − | − | + | − | 10/33 | 11/36 | 30.6 |
| Abnormal brain MRI | − | − | − | − | 3/14 | 3/17 | 17.6 |
| Muscle tone issue | − | + | − | − | 9/24 | 10/27 | 37.0 |
| Age | Assessment Tool | Score | |||||
|---|---|---|---|---|---|---|---|
| Gross Motor | Fine Motor | Language | Personal–Social | Adaptive Behavior | |||
| Patient 1 | 13 month | CNBS | 64 | 67 | 60 | 60 | 67 |
| 37 month | CNBS | 104 | 72 | 85 | 77 | 77 | |
| Patient 2 | 10 month | ASQ | 5 | 55 | 30 | 25 | 50 |
| 14 month | ASQ | 35 | 45 | 30 | 45 | 50 | |
| Patient 3 | 76 month | CNBS | 59 | 63 | 63 | 65 | 59 |
| Patient 4 | 20 month | CNBS | 62 | 57 | 58 | 56 | 63 |
| 24 month | CNBS | 80 | 62 | 65 | 62 | 69 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Xie, H.; Yang, S.; Shangguan, S.; Wang, J.; Jin, C.; Zhang, Y.; Cui, X.; Lyu, Y.; Chen, X.; et al. Possible Catch-Up Developmental Trajectories for Children with Mild Developmental Delay Caused by NAA15 Pathogenic Variants. Genes 2022, 13, 536. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030536
Tian Y, Xie H, Yang S, Shangguan S, Wang J, Jin C, Zhang Y, Cui X, Lyu Y, Chen X, et al. Possible Catch-Up Developmental Trajectories for Children with Mild Developmental Delay Caused by NAA15 Pathogenic Variants. Genes. 2022; 13(3):536. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030536
Chicago/Turabian StyleTian, Yu, Hua Xie, Shenghai Yang, Shaofang Shangguan, Jianhong Wang, Chunhua Jin, Yu Zhang, Xiaodai Cui, Yanyu Lyu, Xiaoli Chen, and et al. 2022. "Possible Catch-Up Developmental Trajectories for Children with Mild Developmental Delay Caused by NAA15 Pathogenic Variants" Genes 13, no. 3: 536. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030536