
Biallelic Loss of Function Mutation in Sodium Channel Gene SCN10A in an Autism Spectrum Disorder Trio from Pakistan

 , and
, and 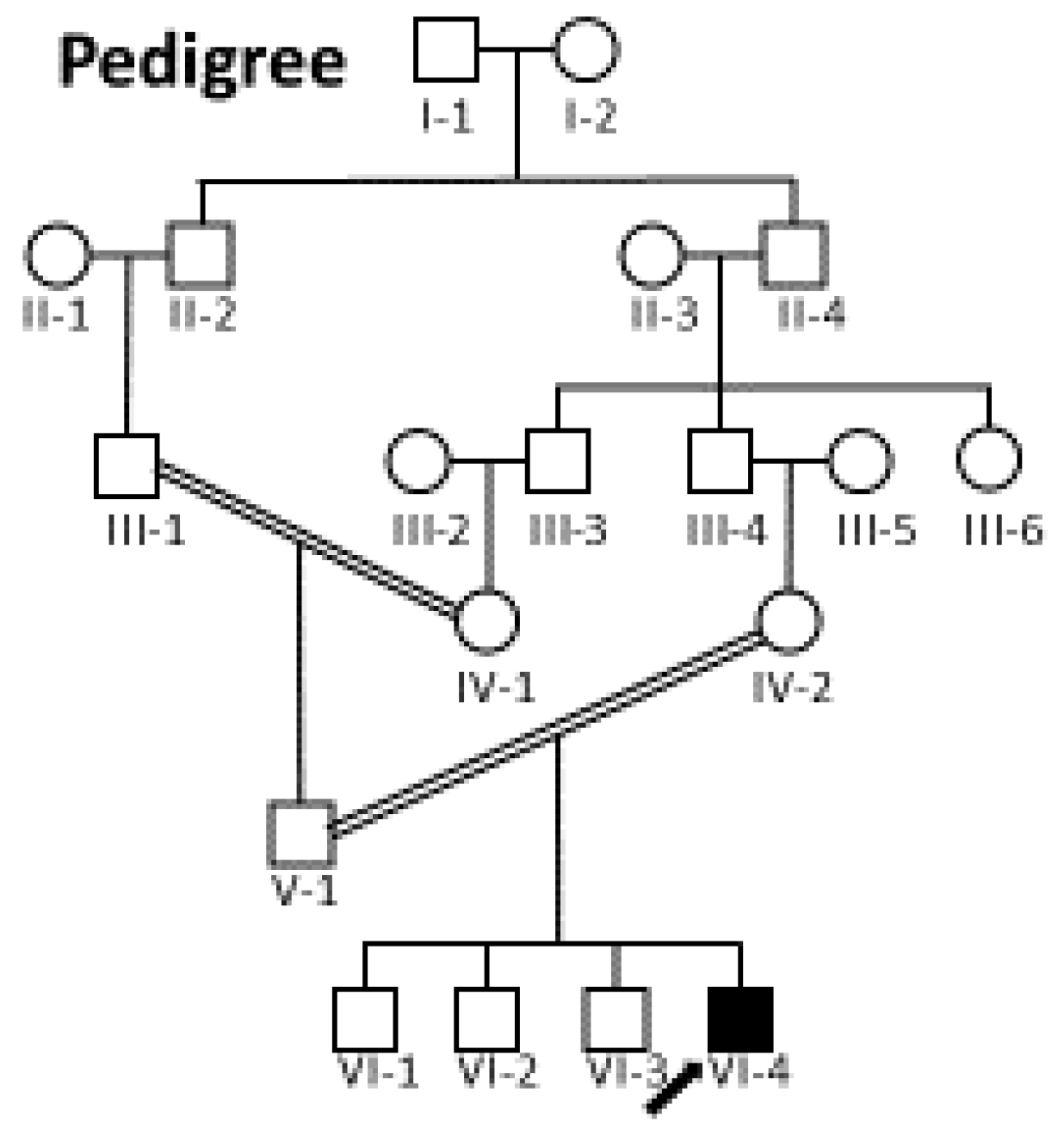{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Ascertainment
2.2. DNA Extraction, Microarray and Sequencing
3. Results
3.1. Clinical Assessment
3.2. Dysmorphology Examination (Done at the Age of 4.4 Years)
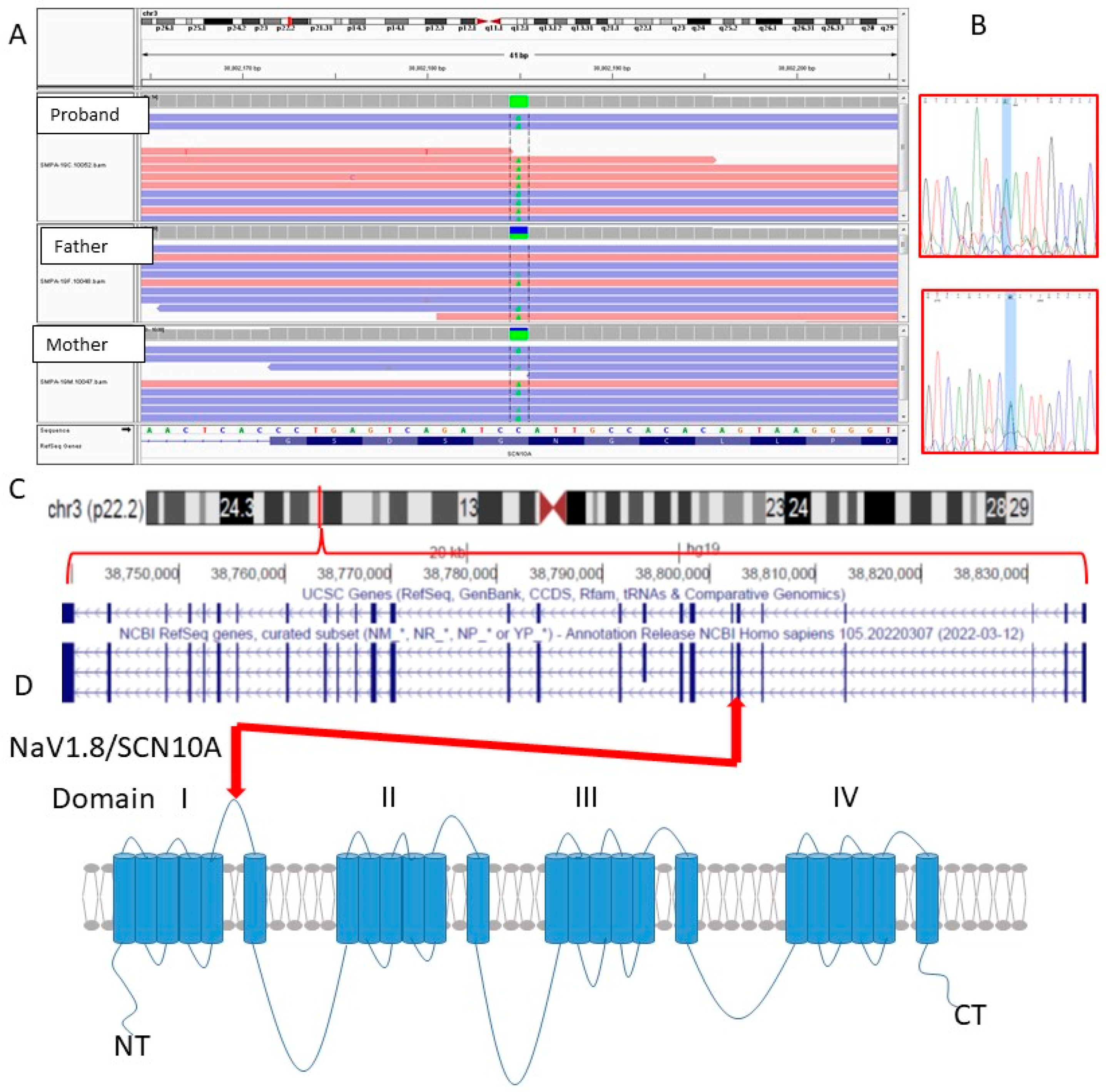
3.3. Genomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| ASD | Autism Spectrum Disorders |
| FEPS2; MIM 615551 | Familial episodic pain syndrome-2 |
| LoF mutation | Loss-of-function |
| BrS | Brugada syndrome |
| MMR) | Measles, Mumps and Rubella |
| M-CHAT | Modified Checklist for Autism in Toddlers |
| CARS | Childhood Autism Rating Scale |
| DSM5 | Diagnostic and Statistical Manual of Mental Disorders of American Psychiatric Association-Fifth Edition |
| SNAP | Sensory nerve action potential |
| MUAPs | Motor unit action potentials |
| CNV | Copy Number Variant |
References
- Faber, C.G.; Lauria, G.; Merkies, I.S.J.; Cheng, X.; Han, C.; Ahn, H.-S.; Persson, A.-K.; Hoeijmakers, J.G.J.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Makiyama, T.; Horie, M. Novel SCN10A variants associated with Brugada syndrome. Europace 2016, 18, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Harripaul, R.; Rabia, A.; Vasli, N.; Mikhailov, A.; Rodrigues, A.; Pastore, S.F.; Muhammad, T.; Madanagopal, T.T.; Hashmi, A.; Tran, C.; et al. Autism spectrum disorder trios from consanguineous populations are enriched for rare biallelic variants, identifying 32 new candidate genes. medRxiv 2022. [Google Scholar] [CrossRef]
- Et, A.; Lifton, R.; Sestan, N.; Mane, S.; Bilguvar, K.; Meyer, K.; Carriero, N.; Bjornson, R.; Overton, J.; Choi, M.; et al. De Novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar]
- Tavassoli, T.; Kolevzon, A.; Wang, A.T.; Curchack-Lichtin, J.; Halpern, D.; Schwartz, L.; Soffes, S.; Bush, L.; Grodberg, D.; Cai, G.; et al. De Novo SCN2A splice site mutation in a boy with Autism spectrum disorder. BMC Med. Genet. 2014, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Robins, D.L.; Fein, D.; Barton, M.L.; Green, J.A. The Modified Checklist for Autism in Toddlers: An initial study investigating the early detection of autism and pervasive developmental disorders. J. Autism Dev. Disord. 2001, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Schopler, E.; Reichler, R.J.; Renner, B.R. The Childhood Autism Rating Scale; Western Psychological Services: Los Angeles, CA, USA, 1988. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; DSM-5 Task Force; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Heinrichs, B.; Liu, B.; Zhang, J.; Meents, J.E.; Le, K.; Erickson, A.; Hautvast, P.; Zhu, X.; Li, N.; Liu, Y.; et al. The Potential Effect of Nav1.8 in Autism Spectrum Disorder: Evidence from a Congenital Case with Compound Heterozygous SCN10A Mutations. Front. Mol. Neurosci. 2021, 14, 709228. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, J.; Qiao, Y.; Calli, K.; Martell, S.; Race, S.; Chijiwa, C.; Glodjo, A.; Jones, S.; Rajcan-Separovic, E.; Scherer, S.W.; et al. Contribution of Multiple Inherited Variants to Autism Spectrum Disorder (ASD) in a Family with 3 Affected Siblings. Genes 2021, 12, 1053. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Di Cui, D.; Han, L.; Xu, L.; Mao, S.; Yang, C.; Gao, F.; Yuan, Z. A novel SCN9A gene variant identified in a Chinese girl with paroxysmal extreme pain disorder (PEPD): A rare case report. BMC Med. Genom. 2022, 15, 159. [Google Scholar] [CrossRef] [PubMed]
- Marchi, M.; D’Amato, I.; Andelic, M.; Cartelli, D.; Salvi, E.; Lombardi, R.; Gumus, E.; Lauria, G. Congenital insensitivity to pain: A novel mutation affecting a U12-type intron causes multiple aberrant splicing of SCN9A. Pain 2022, 163, e882–e887. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma 2013, 43, 11.10.1–11.10.33. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabia, A.; Harripaul, R.; Mikhailov, A.; Mahmood, S.; Maqbool, S.; Vincent, J.B.; Ayub, M. Biallelic Loss of Function Mutation in Sodium Channel Gene SCN10A in an Autism Spectrum Disorder Trio from Pakistan. Genes 2022, 13, 1633. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091633
Rabia A, Harripaul R, Mikhailov A, Mahmood S, Maqbool S, Vincent JB, Ayub M. Biallelic Loss of Function Mutation in Sodium Channel Gene SCN10A in an Autism Spectrum Disorder Trio from Pakistan. Genes. 2022; 13(9):1633. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091633
Chicago/Turabian StyleRabia, Ansa, Ricardo Harripaul, Anna Mikhailov, Saqib Mahmood, Shazia Maqbool, John B. Vincent, and Muhammad Ayub. 2022. "Biallelic Loss of Function Mutation in Sodium Channel Gene SCN10A in an Autism Spectrum Disorder Trio from Pakistan" Genes 13, no. 9: 1633. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091633