
Genomic Diversity and Runs of Homozygosity in Bernese Mountain Dogs
 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Whole-Genome Sequence Data
2.3. Population Structure
2.4. Runs of Homozygosity
3. Results
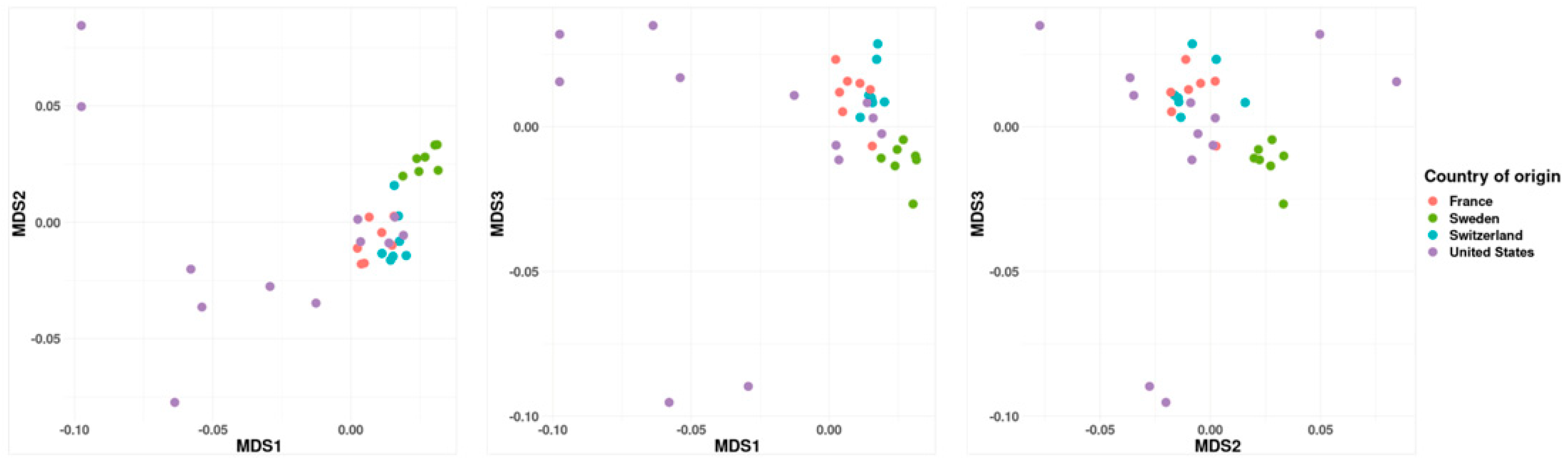
3.1. Population Structure
3.2. Runs of Homozygosity
3.3. ROH Islands
3.4. Distribution of Known Trait-Associated Variants
3.5. Mitochondrial Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fédération Cynologique Internationale. Available online: http://www.fci.be/en/ (accessed on 1 June 2022).
- Räber, H. Die Schweizer Hunderassen; Schweizerische Kynologische Gesellschaft SKG: Bern, Switzerland, 2008; ISBN 978-3-033-01523-4. [Google Scholar]
- Nielsen, L.; Andreasen, S.N.; Andersen, S.D.; Kristensen, A.T. Malignant Histiocytosis and Other Causes of Death in Bernese Mountain Dogs in Denmark. Vet. Rec. 2010, 166, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, M.; Howard, J.; Rossetti, M.; Geissbühler, U. Life Expectancy and Causes of Death in Bernese Mountain Dogs in Switzerland. BMC Vet. Res. 2016, 12, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathmann, I.; Jaggy, A.; Busato, A.; Bärtschi, M.; Gaillard, C. Clinical and Genetic Investigations of Idiopathic Epilepsy in the Bernese Mountain Dog. J. Small Anim. Pract. 1999, 40, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Wininger, F.A.; Zeng, R.; Johnson, G.S.; Katz, M.L.; Johnson, G.C.; Bush, W.W.; Jarboe, J.M.; Coates, J.R. Degenerative Myelopathy in a Bernese Mountain Dog with a Novel SOD1 Missense Mutation. J. Vet. Intern. Med. 2011, 25, 1166–1170. [Google Scholar] [CrossRef]
- Ohlerth, S.; Geiser, B.; Flückiger, M.; Geissbühler, U. Prevalence of Canine Hip Dysplasia in Switzerland Between 1995 and 2016—A Retrospective Study in 5 Common Large Breeds. Front. Vet. Sci. 2019, 6, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hédan, B.; Cadieu, É.; Rimbault, M.; Vaysse, A.; de Citres, C.D.; Devauchelle, P.; Botherel, N.; Abadie, J.; Quignon, P.; Derrien, T.; et al. Identification of Common Predisposing Loci to Hematopoietic Cancers in Four Dog Breeds. PLoS Genet 2021, 17, e1009395. [Google Scholar] [CrossRef]
- Abadie, J.; Hédan, B.; Cadieu, E.; de Brito, C.; Devauchelle, P.; Bourgain, C.; Parker, H.G.; Vaysse, A.; Margaritte-Jeannin, P.; Galibert, F.; et al. Epidemiology, Pathology, and Genetics of Histiocytic Sarcoma in the Bernese Mountain Dog Breed. J. Hered. 2009, 100, S19–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeckx, B.J.G. The Dog 2.0: Lessons Learned from the Past. Theriogenology 2020, 150, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccardo, A.; Marelli, S.P.; Pravettoni, D.; Bagnato, A.; Busca, G.A.; Strillacci, M.G. The German Shorthair Pointer Dog Breed (Canis Lupus Familiaris): Genomic Inbreeding and Variability. Animals 2020, 10, 498. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Biscarini, F.; Tolone, M.; Auzino, B.; Ragatzu, M.; Spaterna, A.; Ciampolini, R. Genomic Characterization of the Braque Français Type Pyrénées Dog and Relationship with Other Breeds. PLoS ONE 2018, 13, e0208548. [Google Scholar] [CrossRef] [Green Version]
- Mortlock, S.A.; Khatkar, M.S.; Williamson, P. Comparative Analysis of Genome Diversity in Bullmastiff Dogs. PLoS ONE 2016, 11, e0147941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, P.X.Y.; Hsu, W.T.; Khatkar, M.S.; Williamson, P. Evaluation of Genetic Diversity and Management of Disease in Border Collie Dogs. Sci. Rep. 2021, 11, 6243. [Google Scholar] [CrossRef]
- Friedenberg, S.G.; Meurs, K.M.; Mackay, T.F.C. Evaluation of Artificial Selection in Standard Poodles Using Whole-Genome Sequencing. Mamm. Genome 2016, 27, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Metzger, J.; Pfahler, S.; Distl, O. Variant Detection and Runs of Homozygosity in next Generation Sequencing Data Elucidate the Genetic Background of Lundehund Syndrome. BMC Genom. 2016, 17, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, A.; Minor, K.M.; Jagannathan, V.; Seefried, F.R.; Mickelson, J.R.; Oliehoek, P.; Drögemüller, C. Genomic Diversity and Population Structure of the Leonberger Dog Breed. Genet. Sel. Evol. 2020, 52, 61. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Botherel, N.; Cadieu, E.; Lagoutte, L.; Hédan, B.; Garand, A.; Abadie, J.; Tiret, L.; Abitbol, M.; Lavoué, R.; et al. Cani-DNA, Un CRB Qui a Du Chien ! Réseau de Collecte de Prélèvements de Chiens Par Les Vétérinaires Pour La Recherche Biomédicale et La Diversité Génétique. NOV’AE 2022, #02, 34–44. [Google Scholar] [CrossRef]
- Ostrander, E.A.; Wang, G.-D.; Larson, G.; VonHoldt, B.M.; Davis, B.W.; Jagannathan, V.; Hitte, C.; Wayne, R.K.; Zhang, Y.-P.; André, C.; et al. Dog10K: An International Sequencing Effort to Advance Studies of Canine Domestication, Phenotypes and Health. Natl. Sci. Rev. 2019, 6, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, V.; Drögemüller, C.; Leeb, T. Dog Biomedical Variant Database Consortium, (DBVDC) A Comprehensive Biomedical Variant Catalogue Based on Whole Genome Sequences of 582 Dogs and Eight Wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wang, C.; Wallerman, O.; Arendt, M.-L.; Sundström, E.; Karlsson, Å.; Nordin, J.; Mäkeläinen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, Å.; et al. A Novel Canine Reference Genome Resolves Genomic Architecture and Uncovers Transcript Complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE 33rd International Parallel and Distributed Processing Symposium, IPDPS 2019, Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Pereira, L.; Van Asch, B.; Amorim, A. Standardisation of Nomenclature for Dog MtDNA D-Loop: A Prerequisite for Launching a Canis Familiaris Database. Forensic. Sci. Int. 2004, 141, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Duleba, A.; Skonieczna, K.; Bogdanowicz, W.; Malyarchuk, B.; Grzybowski, T. Complete Mitochondrial Genome Database and Standardized Classification System for Canis Lupus Familiaris. Forensic. Sci. Int. Genet. 2015, 19, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 6 October 2022).
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to Study Runs of Homozygosity Using Plink? A Guide for Analyzing Medium Density Snp Data in Livestock and Pet Species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of Homozygosity Reveal Highly Penetrant Recessive Loci in Schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of Homozygosity: Windows into Population History and Trait Architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A Publicly Available Repository of ROH Islands Reveals Signatures of Selection in Different Livestock and Pet Species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Hédan, B. Molecular Basis of Canine Histiocytic Sarcoma in Dogs. Available online: https://berner-iwg.org/wp-content/uploads/2022/10/6-Dr-Benoit-Hedan-IWG-2022.pdf (accessed on 6 October 2022).
- Boyko, A.R.; Quignon, P.; Li, L.; Schoenebeck, J.J.; Degenhardt, J.D.; Lohmueller, K.E.; Zhao, K.; Brisbin, A.; Parker, H.G.; vonHoldt, B.M.; et al. A Simple Genetic Architecture Underlies Morphological Variation in Dogs. PLoS Biol. 2010, 8, e1000451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/gene/ (accessed on 25 January 2023).
- Awano, T.; Johnson, G.S.; Wade, C.M.; Katz, M.L.; Johnson, G.C.; Taylor, J.F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; et al. Genome-Wide Association Analysis Reveals a SOD1 Mutation in Canine Degenerative Myelopathy That Resemblesnamyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 2794–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivansson, E.L.; Megquier, K.; Kozyrev, S.V.; Murén, E.; Körberg, I.B.; Swofford, R.; Koltookian, M.; Tonomura, N.; Zeng, R.; Kolicheski, A.L.; et al. Variants within the SP110 Nuclear Body Protein Modify Risk of Canine Degenerative Myelopathy. Proc. Natl. Acad. Sci. USA 2016, 113, E3091–E3100. [Google Scholar] [CrossRef] [Green Version]
- van Poucke, M.; Martlé, V.; van Brantegem, L.; Ducatelle, R.; van Ham, L.; Bhatti, S.; Peelman, L.J. A Canine Orthologue of the Human GFAP c.716G>A (p.Arg239His) Variant Causes Alexander Disease in a Labrador Retriever. Eur. J. Hum. Genet. 2015, 24, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Bannasch, D.L.; Kaelin, C.B.; Letko, A.; Loechel, R.; Hug, P.; Jagannathan, V.; Henkel, J.; Roosje, P.; Hytönen, M.K.; Lohi, H.; et al. Dog Colour Patterns Explained by Modular Promoters of Ancient Canid Origin. Nat. Ecol. Evol. 2021, 5, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Plassais, J.; vonHoldt, B.M.; Parker, H.G.; Carmagnini, A.; Dubos, N.; Papa, I.; Bevant, K.; Derrien, T.; Hennelly, L.M.; Whitaker, D.T.; et al. Natural and Human-Driven Selection of a Single Non-Coding Body Size Variant in Ancient and Modern Canids. Curr. Biol. 2022, 32, 889–897.e9. [Google Scholar] [CrossRef] [PubMed]
- Housley, D.J.E.; Venta, P.J. The Long and the Short of It: Evidence That FGF5 Is a Major Determinant of Canine ‘Hair’-Itability. Anim. Genet. 2006, 37, 309–315. [Google Scholar] [CrossRef]
- Dierks, C.; Mömke, S.; Philipp, U.; Distl, O. Allelic Heterogeneity of FGF5 Mutations Causes the Long-Hair Phenotype in Dogs. Anim. Genet. 2013, 44, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.; Willet, C.E.; Haase, B.; Wade, C.M. Genomic Characterization of External Morphology Traits in Kelpies Does Not Support Common Ancestry with the Australian Dingo. Genes 2019, 10, 337. [Google Scholar] [CrossRef] [Green Version]
- Dreger, D.L.; Rimbault, M.; Davis, B.W.; Bhatnagar, A.; Parker, H.G.; Ostrander, E.A. Whole Genome Sequence, SNP Chips and Pedigree Structure: Building Demographic Profiles in Domestic Dog Breeds to Optimize Genetic Trait Mapping. Dis. Model Mech. 2016, 9, 1445–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannasch, D.; Famula, T.; Donner, J.; Anderson, H.; Honkanen, L.; Batcher, K.; Safra, N.; Thomasy, S.; Rebhun, R. The Effect of Inbreeding, Body Size and Morphology on Health in Dog Breeds. Canine Med. Genet. 2021, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Pfahler, S.; Distl, O. A Massive Reduction of the Genetic Diversity in the Lundehund. Anim. Genet. 2014, 45, 154. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and Runs of Homozygosity: A Possible Solution to an Old Problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Quignon, P.; Herbin, L.; Cadieu, E.; Kirkness, E.F.; Hédan, B.; Mosher, D.S.; Galibert, F.; André, C.; Ostrander, E.A.; Hitte, C. Canine Population Structure: Assessment and Impact of Intra-Breed Stratification on SNP-Based Association Studies. PLoS ONE 2007, 2, e1324. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Coates, J.R.; Johnson, G.C.; Hansen, L.; Awano, T.; Kolicheski, A.; Ivansson, E.; Perloski, M.; Lindblad-Toh, K.; O’Brien, D.P.; et al. Breed Distribution of SOD1 Alleles Previously Associated with Canine Degenerative Myelopathy. J. Vet. Intern. Med. 2014, 28, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrzosek, M.; Giza, E.; Plonek, M.; Podgórski, P.; Vandevelde, M. Alexander Disease in a Dog: Case Presentation of Electrodiagnostic, Magnetic Resonance Imaging and Histopathologic Findings with Review of Literature. BMC Vet. Res. 2015, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearin, A.L.; Hédan, B.; Cadieu, E.; Erich, S.A.; Schmidt, E.V.; Faden, D.L.; Cullen, J.; Abadie, J.; Kwon, E.M.; Gröne, A.; et al. The MTAP-CDKN2A Locus Confers Susceptibility to a Naturally Occurring Canine Cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadomoto, S.; Izumi, K.; Mizokami, A. The CCL20-CCR6 Axis in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 5186. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, J.H.; Park, W.J.; Kim, S. Bioinformatic Exploration for Prognostic Significance of Sphingolipid Metabolism-Related Genes in Invasive Ductal Carcinoma Using the Cancer Genome Atlas Cohort. Int. J. Gen. Med. 2021, 14, 4423–4434. [Google Scholar] [CrossRef]
- Qu, H.; Zhu, Y. SMPDL3B Predicts Poor Prognosis and Contributes to Development of Acute Myeloid Leukemia. Front. Mol. Biosci. 2021, 8, 695601. [Google Scholar] [CrossRef]
- Huang, W.C.; Yen, J.H.; Sung, Y.W.; Tung, S.L.; Chen, P.M.; Chu, P.Y.; Shih, Y.C.; Chi, H.C.; Huang, Y.C.; Huang, S.J.; et al. Novel Function of THEMIS2 in the Enhancement of Cancer Stemness and Chemoresistance by Releasing PTP1B from MET. Oncogene 2022, 41, 997–1010. [Google Scholar] [CrossRef]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole Genome Sequencing of Canids Reveals Genomic Regions under Selection and Variants Influencing Morphology. Nat. Commun. 2019, 10, 1489. [Google Scholar] [CrossRef] [Green Version]
- Imes, D.L.; Wictum, E.J.; Allard, M.W.; Sacks, B.N. Identification of Single Nucleotide Polymorphisms within the MtDNA Genome of the Domestic Dog to Discriminate Individuals with Common HVI Haplotypes. Forensic. Sci. Int. Genet. 2012, 6, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Verscheure, S.; Backeljau, T.; Desmyter, S. Dog Mitochondrial Genome Sequencing to Enhance Dog MtDNA Discrimination Power in Forensic Casework. Forensic. Sci. Int. Genet. 2014, 12, 60–68. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chromosome | Region | Genes Annotated in the Region | |
|---|---|---|---|
| Start | End | ||
| 1 | 55,492,829 | 55,778,313 | CCR6, GPR31, LOC484082, LOC111095971, TTLL2 *, LOC111095973 *, UNC93A *, LOC100855528 *, LOC102153253, LOC111090695 *, LOC111095985 * |
| 2 | 71,838,359 | 71,946,097 | SMPDL3B, RPA2, THEMIS2, LOC119870383, PPP1R8, LOC119870569 |
| 2 | 73,728,360 | 74,031,681 | LDLRAP1, LOC119870404, MACO1, RHCE, TMEM50A, RSRP1, LOC478183, LOC478184, LOC119870544 |
| 6 | 5,429,019 | 5,573,855 | CASTOR2, RCC1L, LOC100682844, LOC119872393 |
| 14 | 15,015,814 | 15,089,249 | ZNF804B, LOC102153442 |
| Trait/ Disorder | OMIA ID | Gene | Variant Region 1 | AlternativeAllele/ Haplotype Frequency | Counts | ||
|---|---|---|---|---|---|---|---|
| Ref/Ref | Ref/Alt | Alt/Alt | |||||
| Degenerative myelopathy | 000263-9615 | SOD1 | chr31:g.27123057G>A | 0.379 | 10 | 21 | 2 |
| chr31:g.27118886A>T | 0.015 | 32 | 1 | 0 | |||
| SP110 | chr25:g.42620442G>A | 0.652 | 4 | 15 | 14 | ||
| chr25:g.42623004C>T | 0.636 | 5 | 14 | 14 | |||
| chr25:g.42628769C>T | 0.167 | 23 | 9 | 1 | |||
| chr25:g.42631341G>A | 0.167 | 23 | 9 | 1 | |||
| chr25:g.42633078T>A | 0.348 | 14 | 15 | 4 | |||
| Alexander disease | 001208-9615 | GFAP | chr9:g.18459694G>A | 0.000 | 33 | 0 | 0 |
| Histiocytic sarcoma | 000620-9615 | risk haplotype | chr5:33Mb 2 | 0.667 | 5 | 12 | 16 |
| risk haplotype | chr11:41Mb 3 | 0.727 | 2 | 14 | 17 | ||
| risk haplotype | chr11:44Mb 4 | 0.621 | 4 | 17 | 12 | ||
| risk haplotype | chr14:11Mb 5 | 0.621 | 3 | 19 | 11 | ||
| Height | 002524-9615 | IGF1 | chr15:g.41511739T>C | 0.030 | 31 | 2 | 0 |
| Ear type | 000319-9615 | MSRB3 | chr10:g.8395407delTTTATTTTAT | 0.970 | 1 | 0 | 32 |
| Hair length | 000439-9615 | FGF5 | chr32:g.35475211G>A | 0.000 | 33 | 0 | 0 |
| chr32:g.35475230_35475231dup | 0.000 | 33 | 0 | 0 | |||
| chr32:g.35475218_35475233del | 0.000 | 33 | 0 | 0 | |||
| chr32:g.35486609A>T | 0.000 | 33 | 0 | 0 | |||
| chr32:g.35494497C>A | 1.000 | 0 | 0 | 33 | |||
| Coat color | 000601-9615 | ASIP | chr24:g.[23863804_23863988del; 23865233_23865267A [4]; 23891659_23891781delins [MT319115.1:424_674] | 1.000 | 0 | 0 | 33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letko, A.; Hédan, B.; Snell, A.; Harris, A.C.; Jagannathan, V.; Andersson, G.; Holst, B.S.; Ostrander, E.A.; Quignon, P.; André, C.; et al. Genomic Diversity and Runs of Homozygosity in Bernese Mountain Dogs. Genes 2023, 14, 650. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030650
Letko A, Hédan B, Snell A, Harris AC, Jagannathan V, Andersson G, Holst BS, Ostrander EA, Quignon P, André C, et al. Genomic Diversity and Runs of Homozygosity in Bernese Mountain Dogs. Genes. 2023; 14(3):650. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030650
Chicago/Turabian StyleLetko, Anna, Benoît Hédan, Anna Snell, Alexander C. Harris, Vidhya Jagannathan, Göran Andersson, Bodil S. Holst, Elaine A. Ostrander, Pascale Quignon, Catherine André, and et al. 2023. "Genomic Diversity and Runs of Homozygosity in Bernese Mountain Dogs" Genes 14, no. 3: 650. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030650