The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Complete Mitochondrial Genome Assembly of G. macromaculatus
2.2. Phylogenetic Analyses and Divergence Time Estimation
2.3. Substitution Rate Estimation and Comparison
3. Results
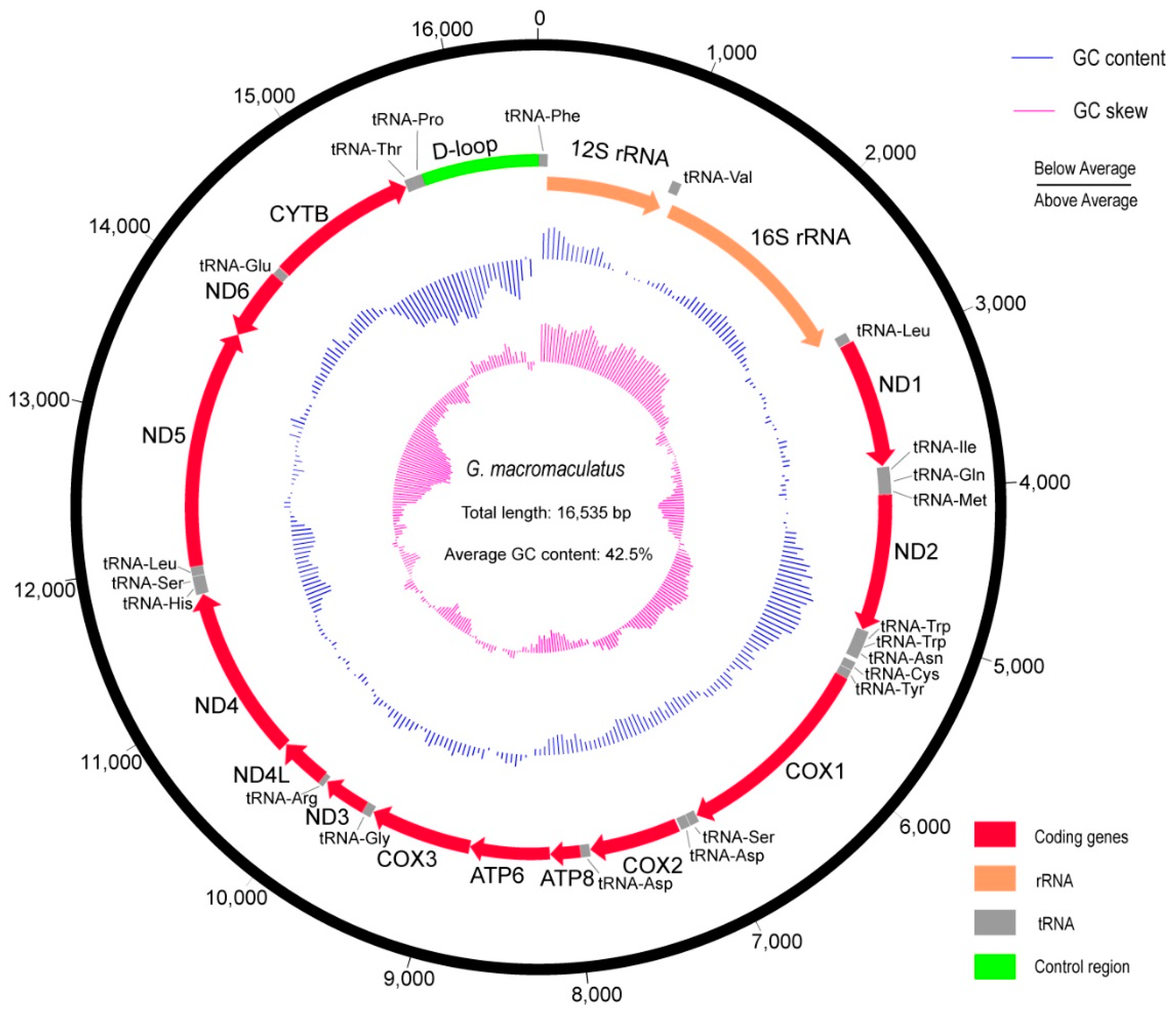
3.1. The Complete Mitochondrial Genome of G. macromaculatus
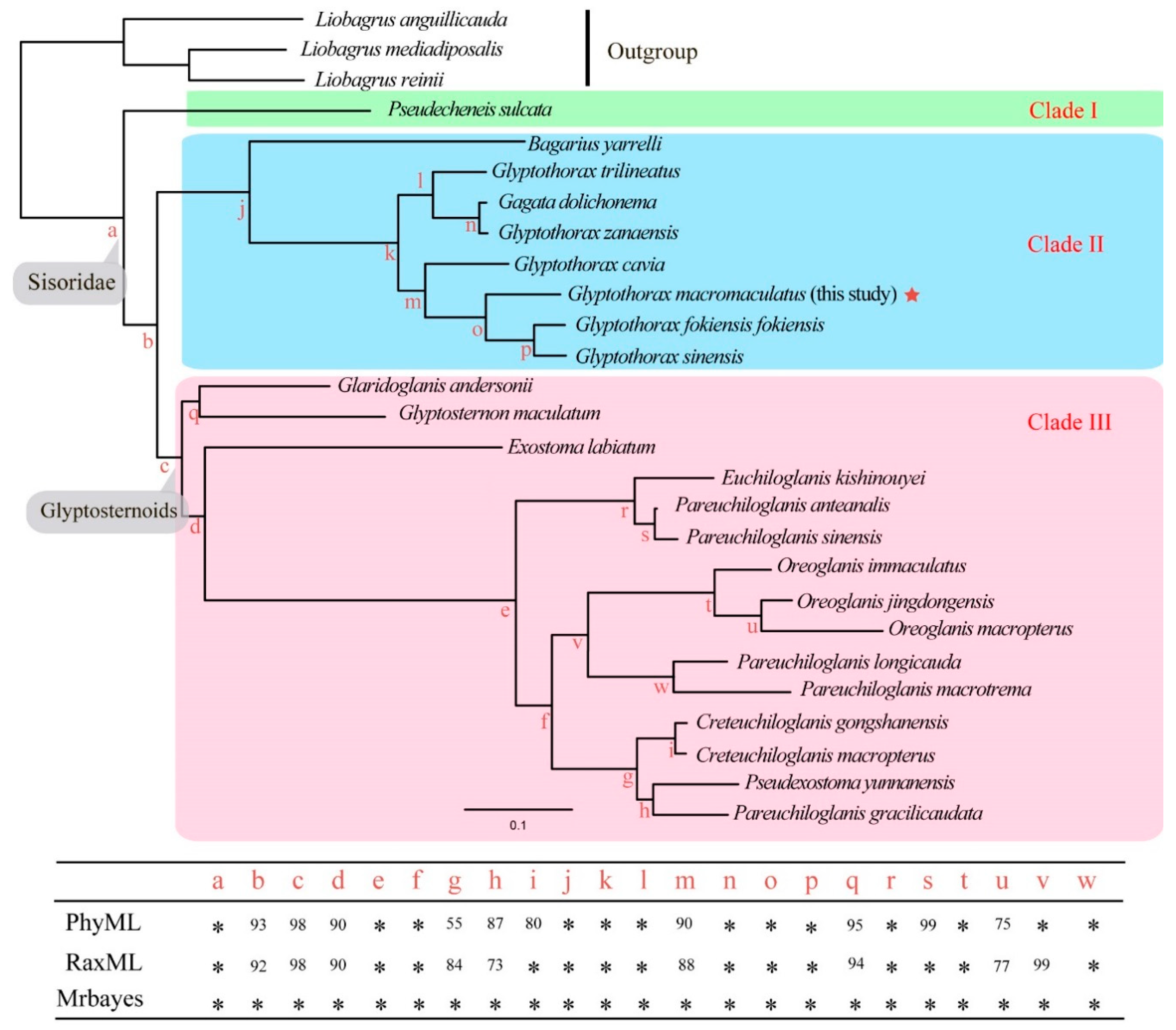
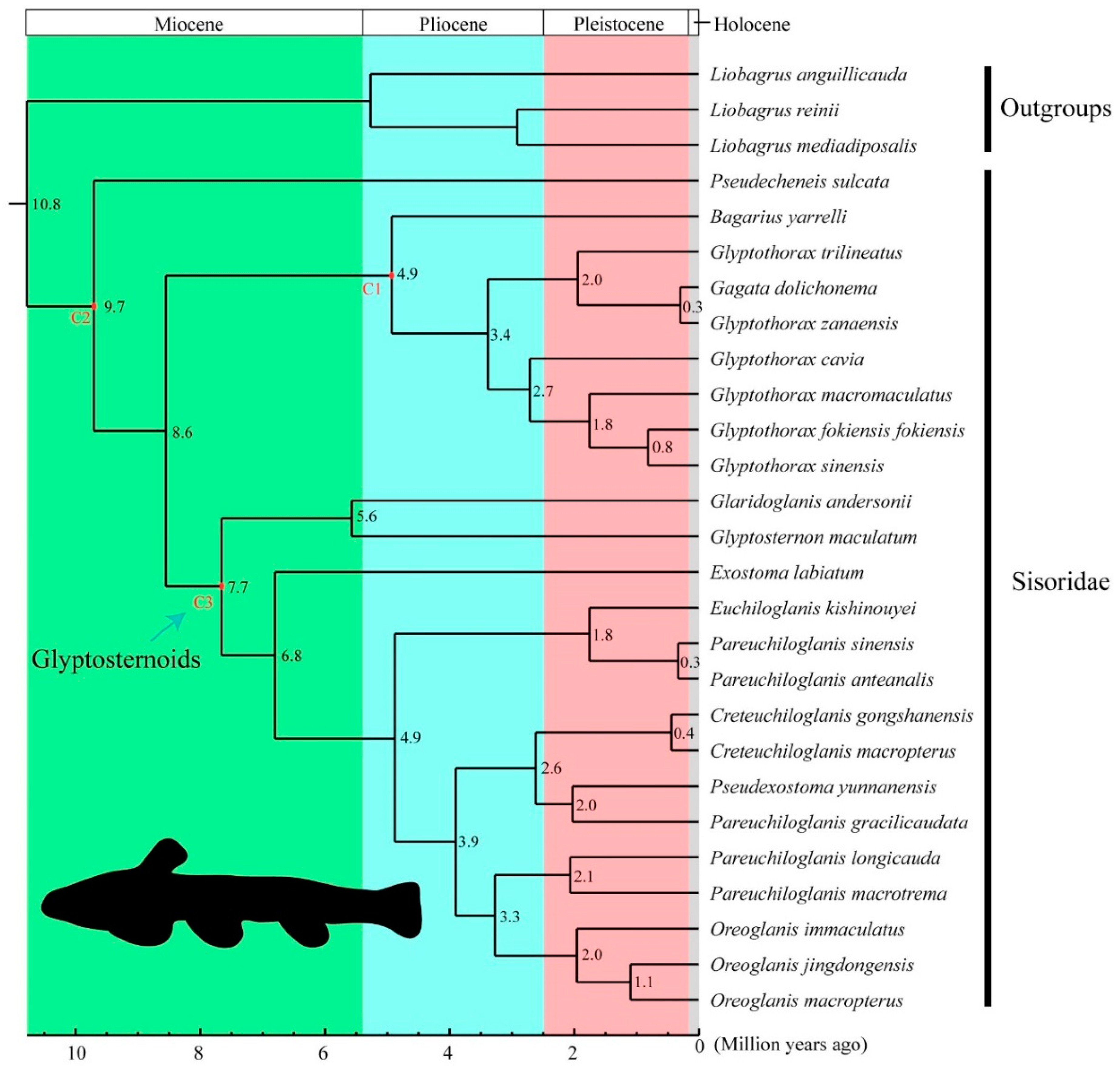
3.2. Phylogenetic Relationships and Divergence Times in the Sisoridae
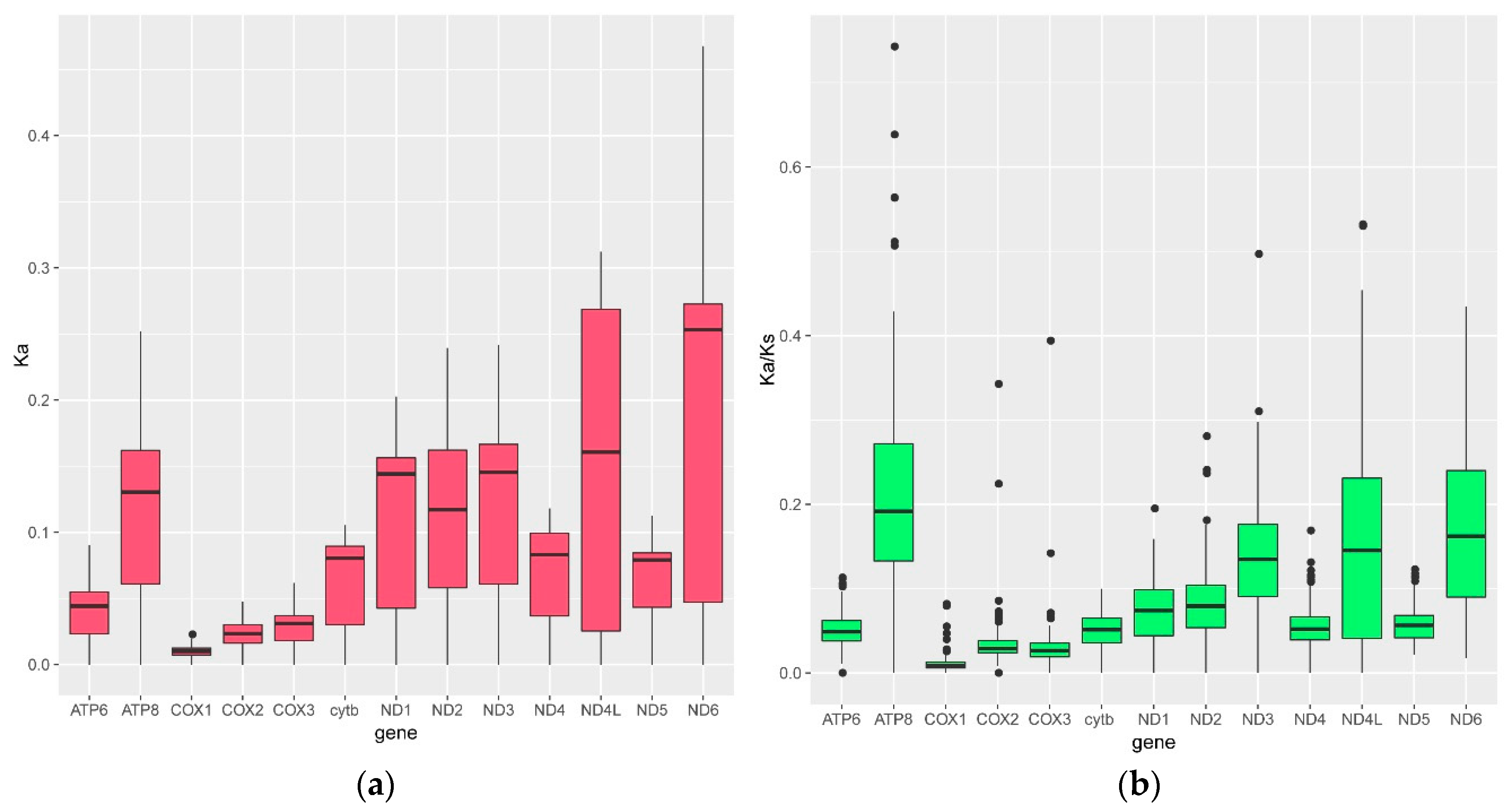
3.3. The Nucleotide Substitution Rate Increased in the Glyptosternoid Fishes
4. Discussion
4.1. Comparison between the Complete Mitochondrial Genomes of G. macromaculatus and Other Vertebrates
4.2. Molecular Phylogeny of the Sisorid Catfishes
4.3. Molecular Dating
4.4. Adaptations to the High Elevation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| atp6 | Adenosine triphosphate synthase subunit 6 |
| atp8 | Adenosine triphosphate synthase subunit 8 |
| bp | Base pair |
| cox1 | Mitochondrial encoded cytochrome c oxidase 1 |
| cox2 | Mitochondrial encoded cytochrome c oxidase 2 |
| cox3 | Mitochondrial encoded cytochrome c oxidase 3 |
| cytb | Cytochrme b |
| Ka | Nonsynonymous substitutions |
| kb | kilobase |
| Ks | Synonymous substitutions |
| nd1 | NADH-Ubiquinone Oxidoreductase Chain 1 |
| nd2 | NADH-Ubiquinone Oxidoreductase Chain 2 |
| nd3 | NADH-Ubiquinone Oxidoreductase Chain 3 |
| nd4 | NADH-Ubiquinone Oxidoreductase Chain 4 |
| nd4l | NADH-Ubiquinone Oxidoreductase Chain 4L |
| nd5 | NADH-Ubiquinone Oxidoreductase Chain 5 |
| ORF | Open reading frame |
| PCR | Polymerase chain reaction |
References
- Hora, S.L.; Silas, E.G. Evolution and distribution of Glyptosternoid fishes of the family Sisoridae (Order: Siluroidea). Proc. Indian Natl. Sci. Acad. B Biol. Sci. 1952, 18, 309–322. [Google Scholar]
- He, S. The phylogeny of the Glyptosternoid fishes (Teleostei: Siluriformes, Sisoridae). Cybium 1996, 20, 115–159. [Google Scholar]
- Guo, X.; Zhang, Y.; He, S.; Chen, Y. Mitochondrial 16s rRNA sequence variations and phylogeny of the Chinese sisorid Catfishes. Chin. Sci. Bull. 2004, 49, 1586–1595. [Google Scholar] [CrossRef]
- Guo, X.; He, S.; Zhang, Y. Phylogeny and biogeography of Chinese sisorid catfishes re-examined using mitochondrial cytochrome b and 16S rRNA gene sequences. Mol. Phylogenet. Evol. 2005, 35, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; He, S.; Zhang, Y. Phylogenetic relationships of Glyptosternoid Fishes (Siluriformes: Sisoridae) inferred from mitochondrial cytochrome b gene sequences. Mol. Phylogenet. Evol. 2004, 31, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; He, S.; Zhang, Y. Phylogenetic relationships of the Chinese sisorid catfishes: A nuclear intron versus mitochondrial gene approach. Hydrobiologia 2007, 579, 55–68. [Google Scholar] [CrossRef]
- Yu, M.; He, S. Phylogenetic relationships and estimation of divergence times among Sisoridae catfishes. Sci. China C Life Sci. 2012, 55, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Ho, S.Y.; Zhang, Y.; He, S. Uplift of the Tibetan Plateau: Evidence from divergence times of Glyptosternoid catfishes. Mol. Phylogenet. Evol. 2006, 39, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, X.; Gang, X.; Zhang, Y.; Irwin, D.M.; Mayden, R.L. Diversification of sisorid catfishes (Teleostei: Siluriformes) in relation to the orogeny of the Himalayan Plateau. Sci. Bull. 2016, 61, 991–1002. [Google Scholar] [CrossRef]
- Ma, X.; Kang, J.; Chen, W.; Zhou, C.; He, S. Biogeographic history and high-elevation adaptations inferred from the mitochondrial genome of Glyptosternoid Fishes (Sisoridae, Siluriformes) from the southeastern Tibetan Plateau. BMC Evol. Biol. 2015, 15, 233. [Google Scholar] [CrossRef] [PubMed]
- Woolfit, M. Effective population size and the rate and pattern of nucleotide substitutions. Biol. Lett. 2009, 5, 417–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohm, J.H.T.; Gwiazdowski, R.A.; Hanner, R. Fast fish face fewer mitochondrial mutations: Patterns of dN/dS across fish mitogenomes. Gene 2015, 572, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Mitterboeck, T.F.; Liu, S.; Adamowicz, S.J.; Fu, J.; Zhang, R.; Song, W.; Meusemann, K.; Zhou, X. Positive and relaxed selection associated with flight evolution and loss in insect transcriptomes. Gigascience 2017, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. Soap2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Lobo, I. Basic local alignment search tool (Blast). J. Mol. Biol. 2012, 215, 403–410. [Google Scholar]
- Mount, D.W. Using the basic local alignment search tool (Blast). Cold Spring Harb. Protoc. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. Mafft version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Phylogenet. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Jmodeltest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O.; Rannala, B. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Kim, I.C.; Jung, S.O.; Lee, Y.M.; Lee, C.J.; Park, J.K.; Lee, J.S. The complete mitochondrial genome of the rayfish Raja porosa (Chondrichthyes, Rajidae). DNA Seq. 2005, 16, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.C.; Kweon, H.S.; Kim, Y.J.; Kim, C.B.; Gye, M.C.; Lee, W.O.; Lee, Y.S.; Lee, J.S. The complete mitochondrial genome of the javeline goby Acanthogobius hasta (Perciformes, Gobiidae) and phylogenetic considerations. Gene 2004, 336, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.C.; Lee, J.S. The complete mitochondrial genome of the rockfish Sebastes schlegeli (Scorpaeniformes, Scorpaenidae). Mol. Cells 2004, 17, 322–328. [Google Scholar] [PubMed]
- Xu, T.J.; Cheng, Y.Z.; Liu, X.Z.; Shi, G.; Wang, R.X. The Complete Mitochondrial genome of the marbled rockfish Sebastiscus marmoratus (Scorpaeniformes, Scorpaenidae): Genome characterization and phylogenetic considerations. Mol. Biol. 2011, 45, 434–445. [Google Scholar] [CrossRef]
- Anderson, S.; de Bruijn, M.H.; Coulson, A.R.; Eperon, I.C.; Sanger, F.; Young, I.G. Complete sequence of bovine mitochondrial DNA conserved features of the mammalian mitochondrial genome. J. Mol. Biol. 1982, 156, 683–717. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; Bruijn, M.H.L.D.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.G.; Masaki, M.; Katsumi, T.; Mutsumi, N. Complete mitochondrial DNA sequence of the Japanese sardine. Fish. Sci. 2001, 67, 828–835. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Chen, S.; Zan, R.; Xiao, H.; Zhang, Y. The complete mitochondrial genomes of two species from Sinocyclocheilus (Cypriniformes: Cyprinidae) and a phylogenetic analysis within Cyprininae. Mol. Biol. Rep. 2010, 37, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Kartavtsev, Y.P.; Jung, S.O.; Lee, Y.M.; Byeon, H.K.; Lee, J.S. Complete mitochondrial genome of the bullhead torrent catfish, Liobagrus Obesus (Siluriformes, Amblycipididae): Genome description and phylogenetic considerations inferred from the Cyt b and 16s rRNA genes. Gene 2007, 396, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.M.; Copeland, P.; Kidd, W.S.; Yin, A. Raising Tibet. Science 1992, 255, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, R.; An, Z. Tectonic uplift in the northern Tibetan Plateau since 13.7 Ma ago inferred from molasse deposits along the altyn tagh fault. Earth Planet. Sci. Lett. 2005, 235, 641–653. [Google Scholar] [CrossRef]
- Clark, M.K.; House, M.A.; Royden, L.H.; Whipple, K.X.; Burchfiel, B.C.; Zhang, X.; Tang, W. Late Genozoic uplift of southeastern Tibet. Geology 2005, 33, 525–528. [Google Scholar] [CrossRef]
- Sun, Y.B.; Shen, Y.Y.; Irwin, D.M.; Zhang, Y. Evaluating the roles of energetic functional constraints on teleost mitochondrial-encoded protein evolution. Mol. Biol. Evol. 2011, 28, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.E.; Li, Q.; Kong, L.; Yu, H. Limited locomotive ability relaxed selective constraints on molluscs mitochondrial genomes. Sci. Rep. 2017, 7, 10628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Xiang, C.; Qi, W.; Xia, S.; Tu, F.; Zhang, X.; Moermond, T.; Yue, B. Phylogenetic analyses and improved resolution of the family Bovidae based on complete mitochondrial genomes. Biochem. Syst. Ecol. 2013, 48, 136–143. [Google Scholar] [CrossRef]
- Xu, S.Q.; Yang, Y.Z.; Zhou, J.; Jing, G.E.; Chen, Y.T.; Wang, J.; Yang, H.M.; Wang, J.; Yu, J.; Zheng, X.G. A mitochondrial genome sequence of the Tibetan antelope (Pantholops hodgsonii). Genom. Proteom. Bioinform. 2005, 3, 5–17. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, Y.; Liu, F.; Gao, Y. Mitochondrial genome of Tibetan wild ass (Equus kiang) reveals substitutions in Nadh which may reflect evolutionary adaption to cold and hypoxic conditions. Asia Life Sci. 2011, 21, 1–11. [Google Scholar]
- Xu, S.; Luosang, J.; Hua, S.; He, J.; Ciren, A.; Wang, W.; Tong, X.; Liang, Y.; Wang, J.; Zheng, X. High altitude adaptation and phylogenetic analysis of Tibetan horse based on the mitochondrial genome. J. Genet. Genom. 2007, 34, 720–729. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, W.; Gao, Y.; Tang, S.; Huang, Q.; Tan, X.; Chen, J.; Huang, T. Mitochondrial genome analysis of ochotona curzoniae and implication of Cytochrome C oxidase in hypoxic adaptation. Mitochondrion 2008, 8, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, Z.; Shedlock, A.M.; Wu, J.; Sang, L.; Tersing, T.; Hasegawa, M.; Yonezawa, T.; Zhong, Y. High altitude adaptation of the schizothoracine fishes (Cyprinidae) revealed by the mitochondrial genome analyses. Gene 2013, 517, 169–178. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Codon | |||||||
|---|---|---|---|---|---|---|---|
| Gene/Element | from | to | Length (bp) | Start | Stop | Intergenic Nucleotides | Strand |
| tRNA-Phe | 1 | 69 | 69 | H | |||
| 12S rRNA | 70 | 1021 | 952 | 5 | H | ||
| tRNA-Val | 1027 | 1098 | 72 | H | |||
| 16S rRNA | 1099 | 2774 | 1677 | H | |||
| tRNA-Leu (UUR) | 2775 | 2849 | 76 | H | |||
| nd1 | 2850 | 3824 | 975 | ATG | TAA | 5 | H |
| tRNA-Ile | 3830 | 3901 | 72 | −1 | H | ||
| tRNA-Gln | 3901 | 3971 | 71 | −2 | L | ||
| tRNA-Met | 3970 | 4038 | 69 | 2 | H | ||
| nd2 | 4041 | 5085 | 1045 | ATG | T-- | H | |
| tRNA-Trp | 5086 | 5156 | 71 | 2 | H | ||
| tRNA-Ala | 5159 | 5227 | 69 | 1 | L | ||
| tRNA-Asn | 5229 | 5301 | 73 | ||||
| tRNA-Cys | 5334 | 5396 | 63 | 6 | L | ||
| tRNA-Tyr | 5403 | 5471 | 69 | 1 | L | ||
| cox1 | 5473 | 7023 | 1551 | GTG | TAA | 1 | H |
| tRNA-Ser (UCN) | 7025 | 7093 | 69 | 5 | L | ||
| tRNA-Asp | 7099 | 7170 | 72 | 15 | H | ||
| cox2 | 7186 | 7876 | 691 | ATG | T-- | H | |
| tRNA-Lys | 7877 | 7950 | 74 | 1 | H | ||
| atp8 | 7952 | 8119 | 168 | ATG | TAA | −8 | H |
| atp6 | 8110 | 8793 | 684 | ATG | TAA | −1 | H |
| cox3 | 8793 | 9576 | 784 | ATG | T-- | H | |
| tRNA-Gly | 9577 | 9648 | 72 | H | |||
| nd3 | 9649 | 9997 | 349 | ATG | T-- | H | |
| tRNA-Arg | 9998 | 10,067 | 70 | H | |||
| nd4l | 10,068 | 10,359 | 292 | ATG | T-- | −2 | H |
| nd4 | 10,358 | 11,738 | 1381 | ATG | T-- | H | |
| tRNA-His | 11,739 | 11,808 | 70 | H | |||
| tRNA-Ser (AGY) | 11,809 | 11,876 | 68 | 5 | H | ||
| tRNA-Leu (CUN) | 11,882 | 11,954 | 73 | H | |||
| nd5 | 11,955 | 13,777 | 1823 | ATG | TAA | H | |
| nd6 | 13,778 | 14,296 | 519 | ATG | TAA | L | |
| tRNA-Glu | 14,297 | 14,365 | 69 | 5 | L | ||
| cytb | 14,371 | 15,508 | 1138 | ATG | T-- | H | |
| tRNA-Thr | 15,509 | 15,576 | 68 | 2 | H | ||
| tRNA-Pro | 15,579 | 15,648 | 70 | L | |||
| D-loop | 15,649 | 16,535 | 887 | - | |||
| Gene | ω0 | ω1 | ω2 | p Value |
|---|---|---|---|---|
| atp6 | 0.03793 | 0.03687 | 2.4219 | 0.009 * |
| atp8 | 0.13819 | 0.13814 | 0.13939 | 0.13 |
| cox1 | 0.01162 | 0.01182 | 0.0001 | 0.15 |
| cox2 | 0.02230 | 0.02230 | 0.0001 | 0.14 |
| cox3 | 0.02792 | 0.02825 | 0.0001 | 0.18 |
| cytb | 0.03201 | 0.03230 | 0.01563 | 0.44 |
| nd1 | 0.05152 | 0.05250 | 0.00927 | 0.08 |
| nd2 | 0.6942 | 0.07010 | 0.03519 | 0.36 |
| nd3 | 0.6969 | 0.6920 | +∞ | 0.27 |
| nd4 | 0.4179 | 0.04198 | 0.03187 | 0.73 |
| nd4l | 0.07551 | 0.07824 | 0.0001 | 0.11 |
| nd5 | 0.05333 | 0.5386 | 0.1308 | 0.20 |
| nd6 | 0.07424 | 0.07390 | 0.08247 | 0.88 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, Y.; Li, Y.; Ruan, Z.; Bian, C.; You, X.; Yang, J.; Jiang, W.; Shi, Q. The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes. Genes 2018, 9, 282. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9060282
Lv Y, Li Y, Ruan Z, Bian C, You X, Yang J, Jiang W, Shi Q. The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes. Genes. 2018; 9(6):282. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9060282
Chicago/Turabian StyleLv, Yunyun, Yanping Li, Zhiqiang Ruan, Chao Bian, Xinxin You, Junxing Yang, Wansheng Jiang, and Qiong Shi. 2018. "The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes" Genes 9, no. 6: 282. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9060282