
Whipple Disease Presenting as Isolated Transverse Myelitis with Permanent Neurological Damage in a Patient with Systemic Lupus Erythematosus: A Case Report of a Difficult Diagnosis with a Literature Review
 , , , , and
, , , , and 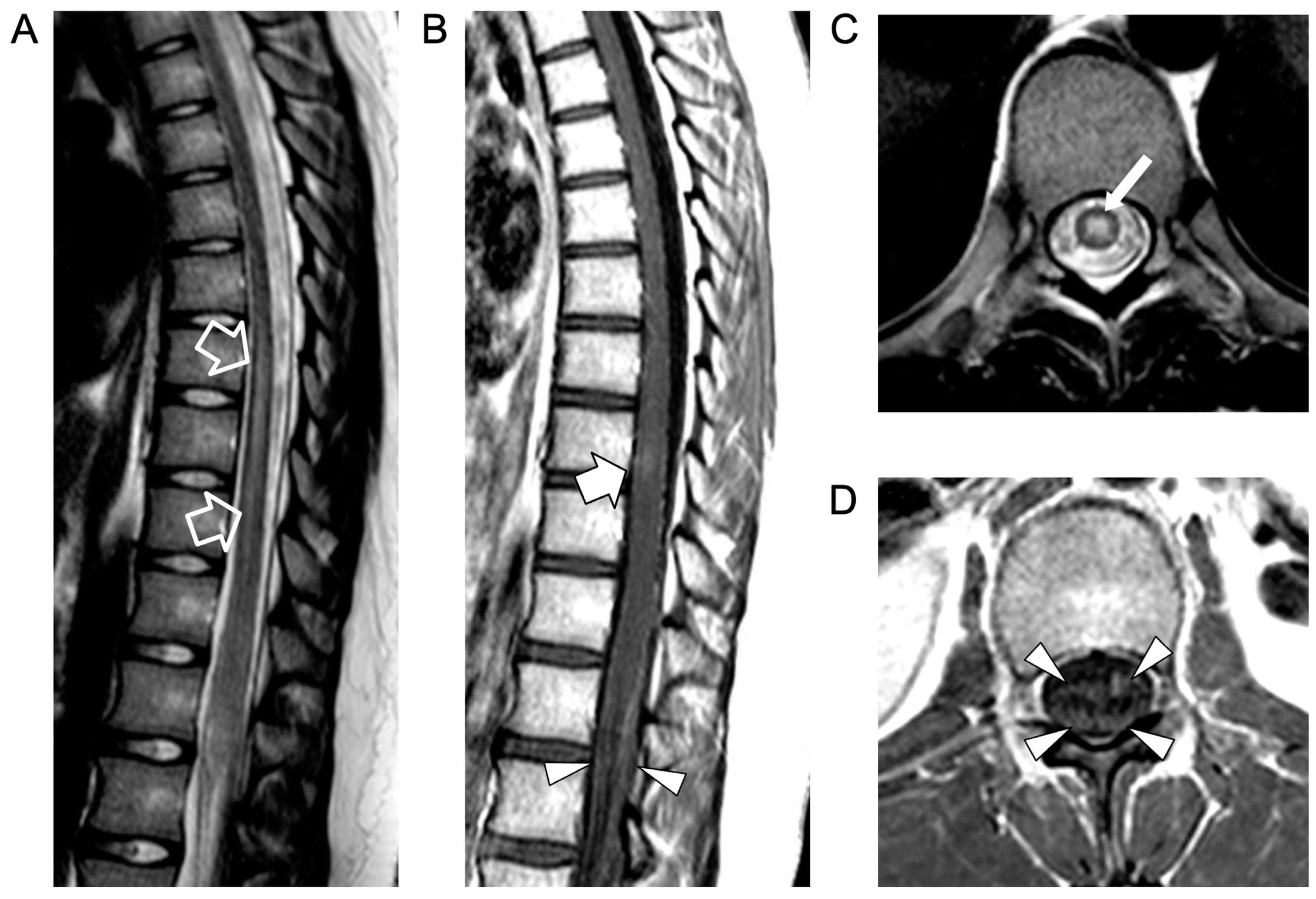{kind=link}
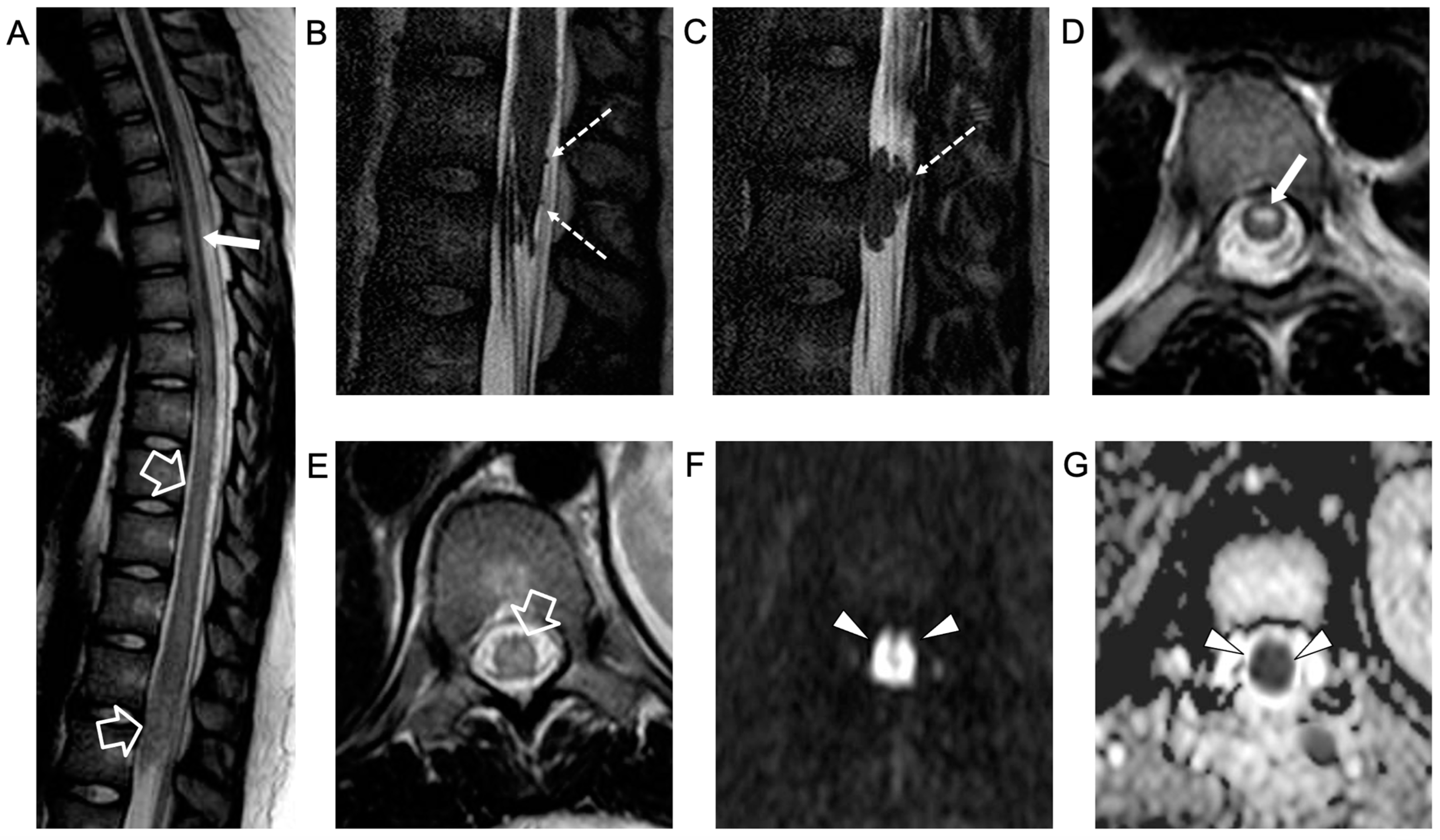{kind=link}
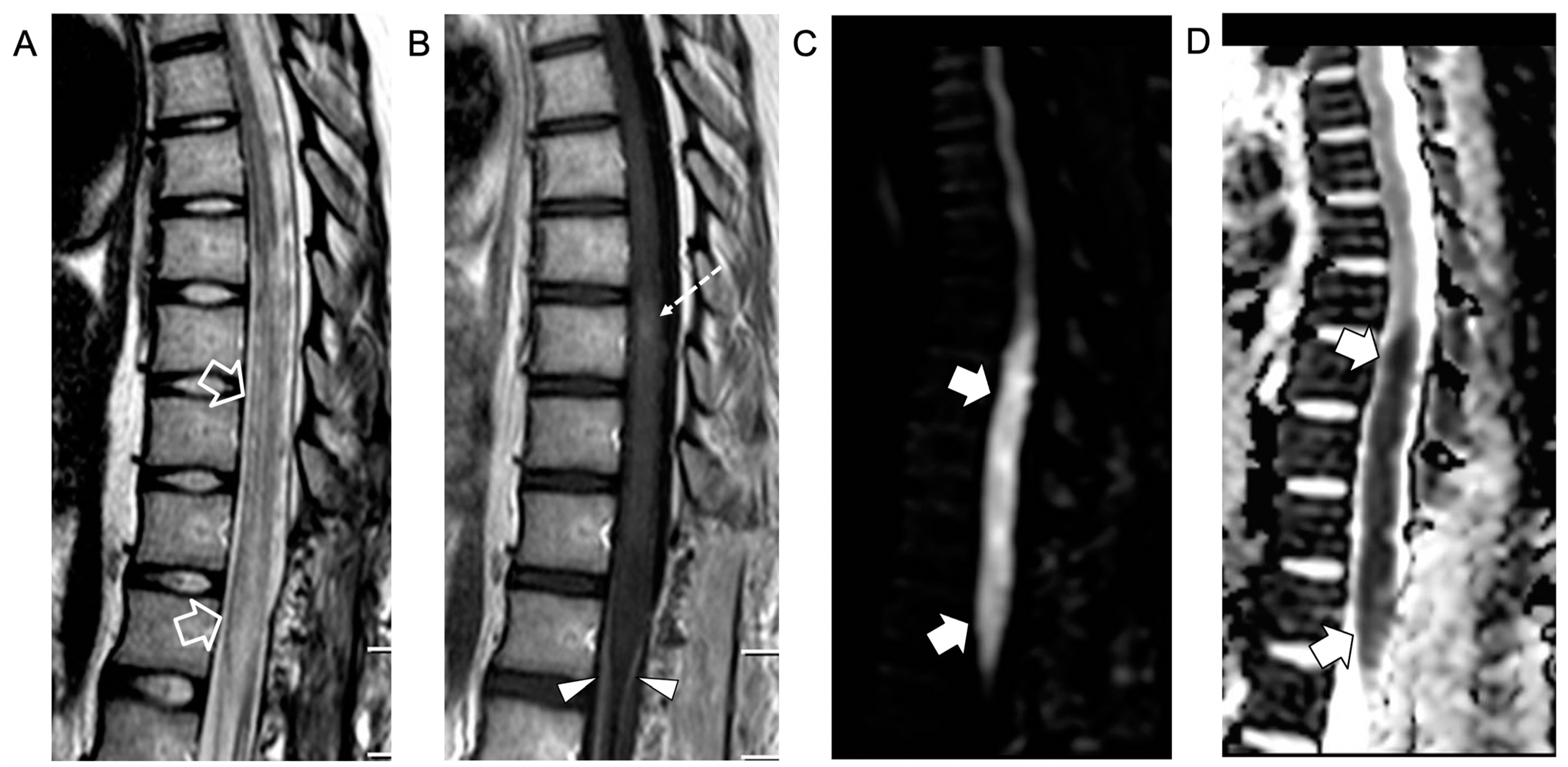{kind=link}
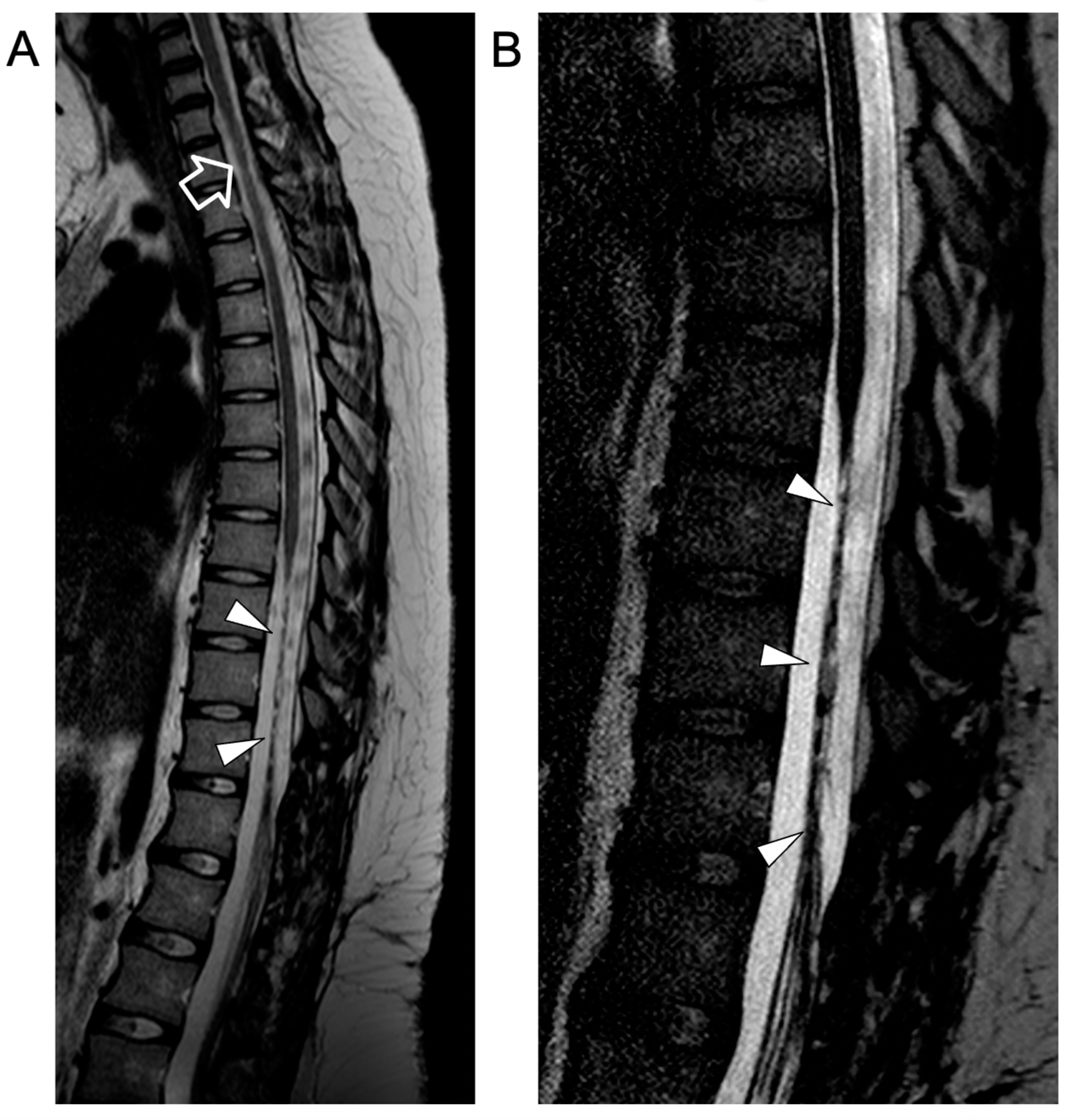{kind=link}
Abstract
:1. Introduction
2. Case Report
3. Literature Review and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marth, T.; Moos, V.; Müller, C.; Biagi, F.; Schneider, T. Tropheryma whipplei infection and Whipple’s disease. Lancet Infect. Dis. 2016, 16, e13–e22. [Google Scholar] [CrossRef] [PubMed]
- El-Abassi, R.; Soliman, M.Y.; Williams, F.; England, J.D. Whipple’s disease. J. Neurol. Sci. 2017, 377, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Compain, C.; Sacre, K.; Puéchal, X.; Klein, I.; Vital-Durand, D.; Houeto, J.L.; De Broucker, T.; Raoult, D.; Papo, T. Central nervous system involvement in whipple disease: Clinical study of 18 patients and long-term follow-up. Medicine 2013, 92, 324–330. [Google Scholar] [CrossRef]
- Jovic, N.S.; Jovic, J.Z. Neurologic disorders in Whipple’s disease. Srp. Arh. Za Celok. Lek. 1996, 124, 98–102. [Google Scholar]
- Pfeiffer, R.F. Gastroenterology and Neurology. Continuum 2017, 23, 744–761. [Google Scholar] [CrossRef]
- Messori, A.; Di Bella, P.; Polonara, G.; Logullo, F.; Pauri, P.; Haghighipour, R.; Salvolini, U. An unusual spinal presentation of Whipple disease. AJNR Am. J. Neuroradiol. 2001, 22, 1004–1008. [Google Scholar]
- El Helou, J.; Saliba, G.; Kolev, I.; Pierrot-Deseilligny, C. Neuro-Whipple confirmed five years after a presumptive diagnosis of a primitive CNS vasculitis. J. Neurol. 2008, 255, 925–926. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.E.; Falope, Z.F.; Abdelhadi, H.A.; Franks, A.J. Cervical myelopathy caused by Whipple’s disease. Neurology 1998, 50, 1505–1506. [Google Scholar] [CrossRef]
- Schroter, A.; Brinkhoff, J.; Gunthner-Lengsfeld, T.; Suerbaum, S.; Reiners, K.; Messmann, H.; Naumann, M. Whipple’s disease presenting as an isolated lesion of the cervical spinal cord. Eur. J. Neurol. 2005, 12, 276–279. [Google Scholar] [CrossRef]
- Pérez Álvarez, Á.I.; Morís de la Tassa, G. Cervical myelopathy as a form of presentation of Whipple disease. Neurologia 2019, 35, 583–585. [Google Scholar] [CrossRef]
- Kremer, S.; Besson, G.; Bonaz, B.; Pasquier, B.; Le Bas, J.F.; Grand, S. Diffuse lesions in the CNS revealed by MR imaging in a case of Whipple disease. AJNR Am. J. Neuroradiol. 2001, 22, 493–495. [Google Scholar]
- Gerard, A.; Sarrot-Reynauld, F.; Liozon, E.; Cathebras, P.; Besson, G.; Robin, C.; Vighetto, A.; Mosnier, J.; Durieu, I.; Vital Durand, D.; et al. Neurologic presentation of whipple disease: Report of 12 cases and review of the literature. Medicine 2002, 81, 443–457. [Google Scholar] [CrossRef]
- Balasa, M.; Gelpi, E.; Rey, M.; Vila, J.; Ramió-Torrentà, L.; Granado, A.M.Q.; Latorre, R.M.; Lepidi, H.; Raoult, D.; Saiz, A. Clinical and Neuropathological Variability in Clinically Isolated Central Nervous System Whipple’s Disease. Brain Pathol. 2014, 24, 230–238. [Google Scholar] [CrossRef]
- Balducci, C.; Foresti, S.; Ciervo, A.; Mancini, F.; Nastasi, G.; Marzorati, L.; Gori, A.; Ferrarese, C.; Appollonio, I.; Peri, A.M. Primary Whipple disease of the Central Nervous System presenting with rhombencephalitis. Int. J. Infect. Dis. 2019, 88, 149–151. [Google Scholar] [CrossRef]
- Ehrenfeld, M.; Urowitz, M.B.; Platts, M.E. Selective C4 deficiency, systemic lupus erythematosus and Whipple’s disease. Ann. Rheum. Dis. 1984, 43, 91–94. [Google Scholar] [CrossRef]
- Losa, F.; Firinu, D.; Deidda, M.; Costanzo, G.; Del Giacco, S.R. Clinical pitfalls of leishmaniasis and Whipple’s disease hidden behind systemic lupus erythematosus: A case series. Acta Microbiol. Immunol. Hung. 2019, 66, 377–385. [Google Scholar] [CrossRef] [PubMed]
- McGlasson, S.; Wiseman, S.; Wardlaw, J.; Dhaun, N.; Hunt, D.P.J. Neurological disease in lupus: Toward a personalized medicine approach. Front. Immunol. 2018, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheumatol. 1999, 42, 599–608. [CrossRef]
- Hanly, J.G.; Urowitz, M.B.; Su, L.; Sanchez-Guerrero, J.; Bae, S.C.; Gordon, C.; Wallace, D.J.; Isenberg, D.; Alarcón, G.S.; Merrill, J.T.; et al. Short-term outcome of neuropsychiatric events in systemic lupus erythematosus upon enrollment into an international inception cohort study. Arthritis Rheumatol. 2008, 59, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Scirè, C.A.; Bombardieri, S.; Caniatti, L.; Conti, F.; De Vita, S.; Doria, A.; Ferraccioli, G.; Gremese, E.; Mansutti, E.; et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology 2015, 54, 891–898. [Google Scholar] [CrossRef]
- Kampylafka, E.I.; Alexopoulos, H.; Kosmidis, M.L.; Panagiotakos, D.B.; Vlachoyiannopoulos, P.G.; Dalakas, M.C.; Moutsopoulos, H.M.; Tzioufas, A.G. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: A 3-year prospective study of 370 patients. PLoS ONE 2013, 8, e55843. [Google Scholar] [CrossRef] [PubMed]
- Dumic, I.; Vitorovic, D.; Spritzer, S.; Sviggum, E.; Patel, J.; Ramanan, P. Acute transverse myelitis—A rare clinical manifestation of Lyme neuroborreliosis. IDCases 2019, 15, e00479. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Wingerchuk, D.M. Clinical practice. Transverse myelitis. N. Engl. J. Med. 2010, 363, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Fenollar, F.; Raoult, D. Acute infections caused by Tropheryma whipplei. Future Microbiol. 2017, 12, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Schöniger-Hekele, M.; Petermann, D.; Weber, B.; Müller, C. Tropheryma whipplei in the environment: Survey of sewage plant influxes and sewage plant workers. Appl. Environ. Microbiol. 2007, 73, 2033–2035. [Google Scholar] [CrossRef]
- Dolmans, R.A.V.; Edwin Boel, C.H.; Lacle, M.M.; Kusters, J.G. Clinical manifestations, treatment, and diagnosis of Tropheryma whipplei infections. Clin. Microbiol. Rev. 2017, 30, 529–555. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Fenollar, F.; Rolain, J.M.; Minodier, P.; Bosdure, E.; Li, W.; Garnier, J.M.; Richet, H. Tropheryma whipplei in children with gastroenteritis. Emerg. Infect. Dis. 2010, 16, 776–782. [Google Scholar] [CrossRef]
- Fenollar, F.; Laouira, S.; Lepidi, H.; Rolain, J.-M.; Raoult, D. Value of Tropheryma whipplei quantitative polymerase chain reaction assay for the diagnosis of Whipple disease: Usefulness of saliva and stool specimens for first-line screening. Clin. Infect. Dis. 2008, 47, 659–667. [Google Scholar] [CrossRef]
- Elchert, J.A.; Mansoor, E.; Abou-Saleh, M.; Cooper, G.S. Epidemiology of Whipple’s Disease in the USA Between 2012 and 2017: A Population-Based National Study. Dig. Dis. Sci. 2019, 64, 1305–1311. [Google Scholar] [CrossRef]
- Moos, V.; Schneider, T. Changing paradigms in Whipple’s disease and infection with Tropheryma whipplei. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1151–1158. [Google Scholar] [CrossRef]
- Fenollar, F.; Keita, A.K.; Buffet, S.; Raoult, D. Intrafamilial circulation of Tropheryma whipplei, France. Emerg. Infect. Dis. 2012, 18, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Keita, A.K.; Brouqui, P.; Badiaga, S.; Benkouiten, S.; Ratmanov, P.; Raoult, D.; Fenollar, F. Tropheryma whipplei prevalence strongly suggests human transmission in homeless shelters. Int. J. Infect. Dis. 2013, 17, e67–e68. [Google Scholar] [CrossRef] [PubMed]
- Edouard, S.; Fenollar, F.; Raoult, D. The rise of Tropheryma whipplei: A 12-year retrospective study of PCR diagnoses in our reference center. J. Clin. Microbiol. 2012, 50, 3917–3920. [Google Scholar] [CrossRef] [PubMed]
- Fenollar, F.; Trani, M.; Davoust, B.; Salle, B.; Birg, M.; Rolain, J.; Raoult, D. Prevalence of asymptomatic Tropheryma whipplei carriage among humans and nonhuman primates. J. Infect. Dis. 2008, 197, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Amsler, L.; Bauerfeind, P.; Nigg, C.; Maibach, R.C.; Steffen, R.; Altwegg, M. Prevalence of Tropheryma whipplei DNA in patients with various gastrointestinal diseases and in healthy controls. Infection 2003, 31, 81–85. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, L.; Pérez-Matute, P.; Blanco, J.R.; Ibarra, V.; Oteo, J.A. High prevalence of asymptomatic carriers of Tropheryma whipplei in different populations from the North of Spain. Enferm. Infecc. Microbiol. Clin. 2016, 34, 340–345. [Google Scholar] [CrossRef]
- Frickmann, H.; Hanke, M.; Hahn, A.; Schwarz, N.G.; Landt, O.; Moter, A.; Kikhney, J.; Hinz, R.; Rojak, S.; Dekker, D.; et al. Detection of Tropheryma whipplei in stool samples by one commercial and two in-house real-time PCR assays. Trop. Med. Int. Health 2019, 24, 101–108. [Google Scholar] [CrossRef]
- Fenollar, F.; Minodier, P.; Boutin, A.; Laporte, R.; Brémond, V.; Noël, G.; Miramont, S.; Richet, H.; Benkouiten, S.; Lagier, J.-C.; et al. Tropheryma whipplei associated with diarrhoea in young children. Clin. Microbiol. Infect. 2016, 22, 869–874. [Google Scholar] [CrossRef]
- Biagi, F.; Balduzzi, D.; Delvino, P.; Schiepatti, A.; Klersy, C.; Corazza, G.R. Prevalence of Whipple’s disease in north-western Italy. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1347–1348. [Google Scholar] [CrossRef]
- Beltrame, A.; Ragusa, A.; Perandin, F.; Formenti, F.; Fenollar, F.; Edouard, S.; Laroche, M.; Zavarise, G.; Doro, F.; Giorli, G.; et al. Tropheryma whipplei intestinal colonization in Italian and migrant population: A retrospective observational study. Future Microbiol. 2019, 14, 283–292. [Google Scholar] [CrossRef]
- Moos, V.; Kunkel, D.; Marth, T.; Feurle, G.E.; LaScola, B.; Ignatius, R.; Zeitz, M.; Schneider, T. Reduced Peripheral and Mucosal Tropheryma whipplei-Specific Th1 Response in Patients with Whipple’s Disease. J. Immunol. 2006, 177, 2015–2022. [Google Scholar] [CrossRef]
- Martinetti, M.; Biagi, F.; Badulli, C.; Feurle, G.E.; Müller, C.; Moos, V.; Schneider, T.; Marth, T.; Marchese, A.; Trotta, L.; et al. The HLA alleles DRB1*13 and DQB1*06 are associated to Whipple’s disease. Gastroenterology 2009, 136, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Mottola, G.; Boucherit, N.; Trouplin, V.; Barry, A.O.; Soubeyran, P.; Mege, J.-L.; Ghigo, E. Tropheryma whipplei, the Agent of Whipple’s Disease, Affects the Early to Late Phagosome Transition and Survives in a Rab5- and Rab7-Positive Compartment. PLoS ONE 2014, 9, e89367. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Schiepatti, A.; Badulli, C.; Sbarsi, I.; Trotta, L.; Feurle, G.E.; Müller, C.; Moos, V.; Schneider, T.; Marth, T.; et al. -295 T-to-C promoter region IL-16 gene polymorphism is associated with Whipple’s disease. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1919–1921. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Fenollar, F.; Raoult, D.; Mege, J.L. Increased Levels of Circulating IL-16 and Apoptosis Markers Are Related to the Activity of Whipple’s Disease. PLoS ONE 2007, 2, 6. [Google Scholar] [CrossRef]
- Biagi, F.; Badulli, C.; Feurle, G.E.; Müller, C.; Moos, V.; Schneider, T.; Marth, T.; Mytilineos, J.; Garlaschelli, F.; Marchese, A.; et al. Cytokine genetic profile in Whipple’s disease. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 3145–3150. [Google Scholar] [CrossRef]
- Kalt, A.; Schneider, T.; Ring, S.; Hoffmann, J.; Zeitz, M.; Stallmach, A.; Persing, D.H.; Marth, T. Decreased levels of interleukin-12p40 in the serum of patients with Whipple’s disease. Int. J. Color. Dis. 2006, 21, 114–120. [Google Scholar] [CrossRef]
- Ben Azzouz, E.; Boumaza, A.; Mezouar, S.; Bardou, M.; Carlini, F.; Picard, C.; Raoult, D.; Mège, J.-L.; Desnues, B. Tropheryma whipplei Increases Expression of Human Leukocyte Antigen-G on Monocytes to Reduce Tumor Necrosis Factor and Promote Bacterial Replication. Gastroenterology 2018, 155, 1553–1563. [Google Scholar] [CrossRef]
- Glaser, C.; Rieg, S.; Wiech, T.; Scholz, C.; Endres, D.; Stich, O.; Hasselblatt, P.; Geißdörfer, W.; Bogdan, C.; Serr, A.; et al. Whipple’s disease mimicking rheumatoid arthritis can cause misdiagnosis and treatment failure. Orphanet J. Rare Dis. 2017, 12, 99. [Google Scholar] [CrossRef]
- Keita, A.K.; Diatta, G.; Ratmanov, P.; Bassene, H.; Raoult, D.; Roucher, C.; Fenollar, F.; Sokhna, C.; Tall, A.; Trape, J.-F.; et al. Looking for Tropheryma whipplei source and reservoir in rural Senegal. Am. J. Trop. Med. Hyg. 2013, 88, 339–343. [Google Scholar] [CrossRef]
- Biagi, F.; Trotta, L.; Corazza, G.R. Whipple’s disease. Intern. Emerg. Med. 2012, 7, 209–213. [Google Scholar] [CrossRef]
- Lagier, J.C.; Raoult, D. Whipple’s disease and Tropheryma whipplei infections: When to suspect them and how to diagnose and treat them. Curr. Opin. Infect. Dis. 2018, 31, 463–470. [Google Scholar] [CrossRef]
- Günther, U.; Moos, V.; Offenmüller, G.; Oelkers, G.; Heise, W.; Moter, A.; Loddenkemper, C.; Schneider, T. Gastrointestinal diagnosis of classical whipple disease: Clinical, endoscopic, and histopathologic features in 191 patients. Medicine 2015, 94, 15. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D. From Whipple Disease to Tropheryma whipplei Infection. In Clinical Infectious Diseases; Oxford University Press: Oxford, UK, 2019; Volume 68, pp. 1098–1099. [Google Scholar]
- Fournier, P.-E.; Gouriet, F.; Casalta, J.-P.; Lepidi, H.; Chaudet, H.; Thuny, F.; Collart, F.; Habib, G.; Raoult, D. Blood culture-negative endocarditis: Improving the diagnostic yield using new diagnostic tools. Medicine 2017, 96, e8392. [Google Scholar] [CrossRef] [PubMed]
- Paymard, M.; Sukumaran, V.; Senanayake, S.; Watson, A.; Das, C.; Abhayaratna, W. Tropheryma Whipplei endocarditis: Case report and literature review. Heart Views 2018, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.C.; Lepidi, H.; Raoult, D.; Fenollar, F. Systemic tropheryma whipplei: Clinical presentation of 142 patients with infections diagnosed or confirmed in a reference center. Medicine 2010, 89, 337–345. [Google Scholar] [CrossRef]
- Marth, T. Complicated Whipple’s disease and endocarditis following tumor necrosis factor inhibitors. World J. Cardiol. 2014, 6, 12. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Fenollar, F.; Lepidi, H.; Giorgi, R.; Million, M.; Raoult, D. Treatment of classic Whipple’s disease: From in vitro results to clinical outcome. J. Antimicrob. Chemother. 2014, 69, 219–227. [Google Scholar] [CrossRef]
- Chandra, S.R.; Raj, P.; Pai, A.R.; Reddy, N. A Case of Whipple’s Disease: A Very Rare Cause for Rapidly Progressive Dementia. Indian J. Psychol. Med. 2019, 40, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Panegyres, P.K.; Goh, J. Sleep disorders of Whipple’s disease of the brain. QJM 2015, 108, 99–103. [Google Scholar] [CrossRef]
- Mohamed, W.; Neil, E.; Kupsky, W.J.; Juhász, C.; Mittal, S.; Santhakumar, S. Isolated inctracranial Whipple’s disease-report of a rare case and review of the literature. J. Neurol. Sci. 2011, 308, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fenollar, F.; Nicoli, F.; Paquet, C.; Lepidi, H.; Cozzone, P.; Antoine, J.-C.; Pouget, J.; Raoult, D. Progressive dementia associated with ataxia or obesity in patients with Tropheryma whipplei encephalitis. BMC Infect. Dis. 2011, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Kilani, M.; Njim, L.; Ben Nsir, A.; Hattab, M.N. Whipple Disease Presenting as Cystic Brain Tumor: Case Report and Review of the Literature. Turk. Neurosurg. 2018, 28, 495–499. [Google Scholar] [PubMed]
- Brönnimann, D.; Vareil, M.-O.; Sibon, I.; Lagier, J.-C.; Lepidi, H.; Puges, M.; Haneche, F.; Raoult, D.; Desclaux, A.; Neau, D.; et al. Limbic encephalitis as a relapse of Whipple’s disease with digestive involvement and spondylodiscitis. Infection 2018, 47, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Seneca, V.; Imperato, A.; Colella, G.; Cioffi, V.; Mariniello, G.; Gangemi, M. Recurrent acute obstructive hydrocephalus as clinical onset of cerebral Whipple’s disease. Clin. Neurol. Neurosurg. 2010, 112, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Mahnel, R.; Kalt, A.; Ring, S.; Stallmach, A.; Strober, W.; Marth, T. Immunosuppressive therapy in Whipple’s disease patients is associated with the appearance of gastrointestinal manifestations. Am. J. Gastroenterol. 2005, 100, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.A.; Moreira, R.K.; Lam-Himlin, D.; De Petris, G.; Montgomery, E. Whipple disease a century after the initial description: Increased recognition of unusual presentations, autoimmune comorbidities, and therapy effects. Am. J. Surg. Pathol. 2012, 36, 1066–1073. [Google Scholar] [CrossRef]
- Biagi, F.; Trotta, L.; Di Stefano, M.; Balduzzi, D.; Marchese, A.; Vattiato, C.; Bianchi, P.I.; Fenollar, F.; Corazza, G.R. Previous immunosuppressive therapy is a risk factor for immune reconstitution inflammatory syndrome in Whipple’s disease. Dig. Liver Dis. 2012, 44, 880–882. [Google Scholar] [CrossRef]
- Feurle, G.E.; Moos, V.; Schinnerling, K.; Geelhaar, A.; Allers, K.; Biagi, F.; Bläker, H.; Moter, A.; Loddenkemper, C.; Jansen, A.; et al. The immune reconstitution inflammatory syndrome in whipple disease: A cohort study. Ann. Intern. Med. 2010, 153, 710–717. [Google Scholar] [CrossRef]
- Lagier, J.C.; Raoult, D. Immune reconstitution inflammatory syndrome associated with bacterial infections. Expert Opin. Drug Saf. 2014, 13, 341–350. [Google Scholar] [CrossRef]
- Schneider, T.; Moos, V.; Loddenkemper, C.; Marth, T.; Fenollar, F.; Raoult, D. Whipple’s disease: New aspects of pathogenesis and treatment. Lancet Infect. Dis. 2008, 8, 179–190. [Google Scholar] [CrossRef]
- Feurle, G.E.; Junga, N.S.; Marth, T. Efficacy of ceftriaxone or meropenem as initial therapies in Whipple’s disease. Gastroenterology 2010, 138, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Moter, A.; Janneck, M.; Wolters, M.; Iking-Konert, C.; Wiessner, A.; Loddenkemper, C.; Hartleben, B.; Lütgehetmann, M.; Schmidt, J.; Langbehn, U.; et al. Potential Role for Urine Polymerase Chain Reaction in the Diagnosis of Whipple’s Disease. Clin. Infect. Dis. 2019, 68, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Tison, A.; Saraux, A. Potential Role for Urine Polymerase Chain Reaction in the Diagnosis of Whipple Disease. Clin. Infect. Dis. 2019, 69, 904–905. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Cammilleri, S.; Raoult, D. Classic Whipple’s disease diagnosed by (18)F-fluorodeoxyglucose PET. Lancet Infect. Dis. 2016, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Tábuas-Pereira, M.; Vicente, M.; Coelho, F.; Santana, I. Prosopagnosia as the presenting symptom of whipple disease. Cogn. Behav. Neurol. 2016, 29, 100–106. [Google Scholar] [CrossRef]
- Peregrin, J.; Malikova, H. Primary Whipple disease of the brain: Case report with long-term clinical and MRI follow-up. Neuropsychiatr. Dis. Treat. 2015, 11, 2461–2469. [Google Scholar]
- Giaccone, G.; Carella, F.; Parravicini, C.; Longhi, E.; Chiapparini, L.; Savoiardo, M.; Montano, N.; Morbin, M.; Albanese, A.; Tagliavini, F. A 52-year-old man with myoclonic jerks. Brain Pathol. 2016, 26, 291–292. [Google Scholar] [CrossRef]
- Feurle, G.E.; Moos, V.; Bläker, H.; Loddenkemper, C.; Moter, A.; Stroux, A.; Marth, T.; Schneider, T. Intravenous ceftriaxone, followed by 12 or three months of oral treatment with trimethoprim-sulfamethoxazole in Whipple’s disease. J. Infect. 2013, 66, 263–270. [Google Scholar] [CrossRef]
- Fenollar, F.; Rolain, J.-M.; Alric, L.; Papo, T.; Chauveheid, M.-P.; van de Beek, D.; Raoult, D. Resistance to trimethoprim/sulfamethoxazole and Tropheryma whipplei. Int. J. Antimicrob. Agents 2009, 34, 255–259. [Google Scholar] [CrossRef]
- Fenollar, F.; Lagier, J.-C.; Raoult, D. Tropheryma whipplei and Whipple’s disease. J. Infect. 2014, 69, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Biagi, G.L.; Corazza, G.R. What is the best therapy for Whipple’s disease? Our point of view. Scand. J. Gastroenterol. 2017, 52, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J. Autoimmun. 2014, 48–49, 10–13. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saffioti, C.; Nebiolo, M.; Caorsi, R.; Mesini, A.; Severino, M.; Brisca, G.; Castagnola, E.; Gattorno, M. Whipple Disease Presenting as Isolated Transverse Myelitis with Permanent Neurological Damage in a Patient with Systemic Lupus Erythematosus: A Case Report of a Difficult Diagnosis with a Literature Review. Infect. Dis. Rep. 2024, 16, 269-280. https://0-doi-org.brum.beds.ac.uk/10.3390/idr16020022
Saffioti C, Nebiolo M, Caorsi R, Mesini A, Severino M, Brisca G, Castagnola E, Gattorno M. Whipple Disease Presenting as Isolated Transverse Myelitis with Permanent Neurological Damage in a Patient with Systemic Lupus Erythematosus: A Case Report of a Difficult Diagnosis with a Literature Review. Infectious Disease Reports. 2024; 16(2):269-280. https://0-doi-org.brum.beds.ac.uk/10.3390/idr16020022
Chicago/Turabian StyleSaffioti, Carolina, Marta Nebiolo, Roberta Caorsi, Alessio Mesini, Mariasavina Severino, Giacomo Brisca, Elio Castagnola, and Marco Gattorno. 2024. "Whipple Disease Presenting as Isolated Transverse Myelitis with Permanent Neurological Damage in a Patient with Systemic Lupus Erythematosus: A Case Report of a Difficult Diagnosis with a Literature Review" Infectious Disease Reports 16, no. 2: 269-280. https://0-doi-org.brum.beds.ac.uk/10.3390/idr16020022