3.2. Molecular Modification of Fluoroquinolone with Low Plasma Protein Binding Rate Based on Contour Maps
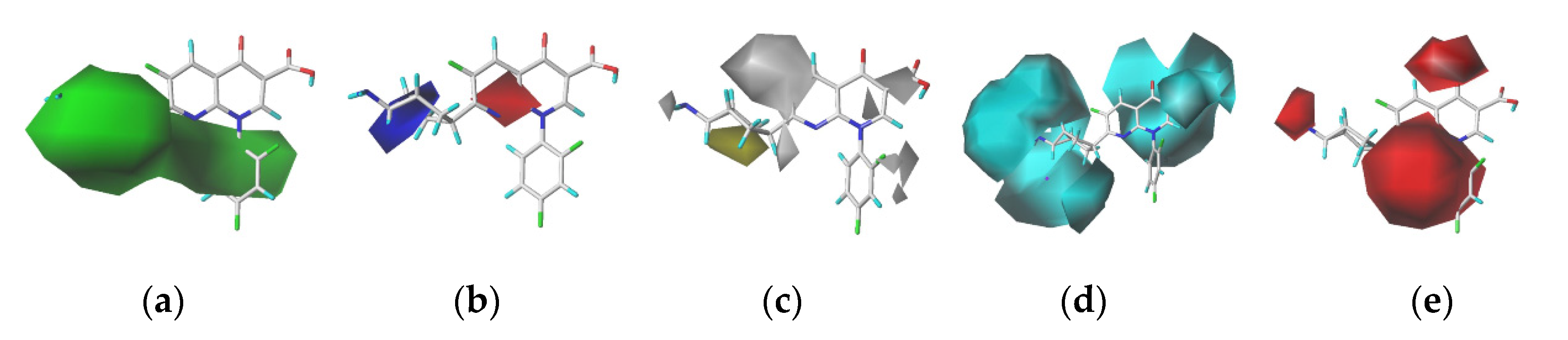
In the CoMSIA model, the contributions of the steric field (S), electrostatic field (E), hydrophobic field (H), hydrogen bond donor field (D), hydrogen bond acceptor field (A) were respectively18.30%, 23.00%, 33.60%, 7.00%, and 18.10%. The results showed that logfb values of quinolones were affected by the spatial effect, electrical distribution, hydrophobicity of groups and hydrogen bond donors and receptors.
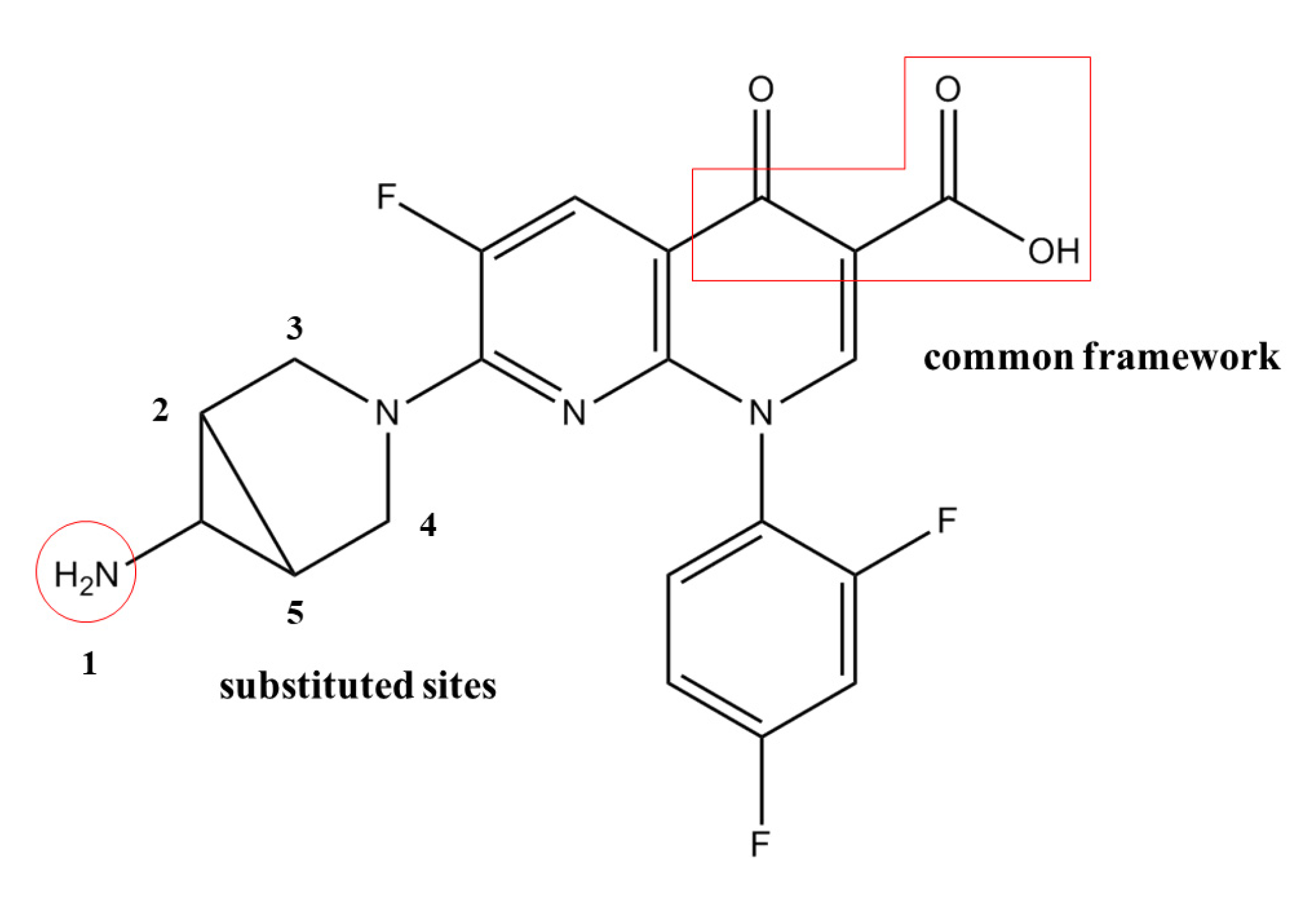
In this study, we selected trovafloxacin as the target molecule. In the contour maps generated using the CoMSIA model (
Figure 2), the steric field represented by the green region is distributed near the 1-, 2-, 3-, 4-, and 5- substituents, indicating that the introduction of small groups at positions 1-, 2-, 3-, 4-, and 5- reduced the log
fb values of the quinolones and reduced their binding rates to plasma protein. The electrostatic field represented by the blue region is distributed near position 5-, indicating that the introduction of electropositive groups at position 5- could reduce the log
fb values of the quinolones. The white region representing the hydrophobic fields was distributed at the 1- substituent, indicating that the introduction of a hydrophobic group at position 1- could reduce the log
fb values of the quinolones. The hydrogen bond donor field represented by the cyan region is distributed at positions 1- and 2-, indicating that the introduction of the hydrogen bond donor at positions 1- and 2- could reduce the log
fb values of the quinolones. The red region representing hydrogen bond acceptors is distributed at the position 1- substituent, indicating that the introduction of the hydrogen bond acceptor at position 1- could reduce the log
fb values of the quinolones.
Comprehensive analysis of the contour maps obtained using the CoMSIA model revealed that position 1- was in the green, white, cyan, and red regions; position 2- was in the green and cyan regions; positions 3- and 4- were in the green region; and position 5- was in the green and blue regions. Position 1- was affected by the steric field, hydrophobic field, and hydrogen bond acceptor field. In other words, the introduction of small, hydrophobic, and hydrogen bond acceptor groups at this position was advantageous for reducing the log
fb values of the quinolones. Position 2- was influenced by the steric and hydrogen bond donor fields, and the introduction of small and hydrogen bond receptor groups at this site could reduce the log
fb values of the quinolones. Positions 3- and 4- were affected by the steric field, and the introduction of small groups at these positions proved beneficial for reducing the log
fb values of the quinolones. The introduction of small and electronegative groups at this position could reduce the log
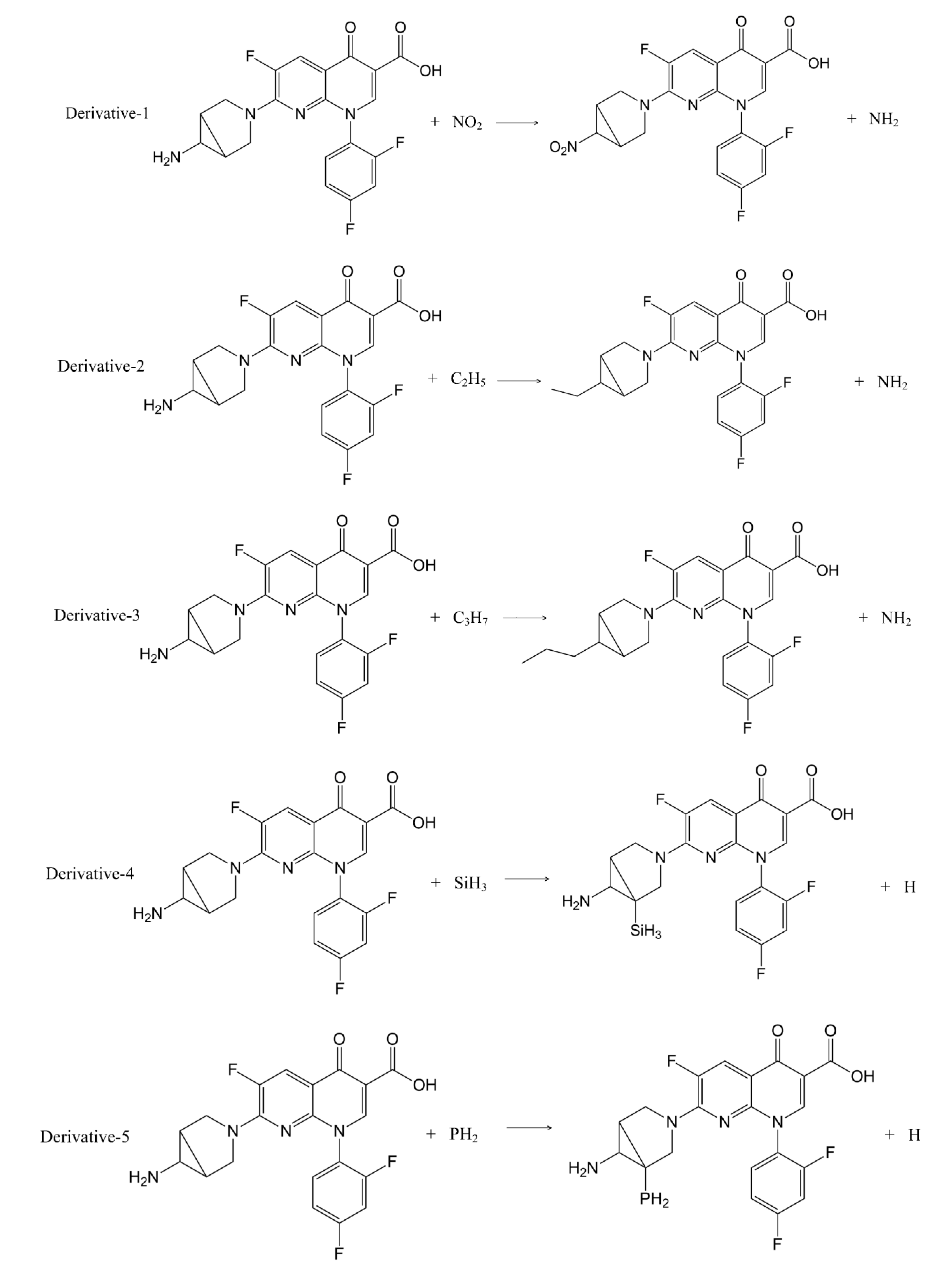
fb values of the quinolones, on account of position 5- was affected by the steric and the electrostatic fields. Therefore, combining with the characteristics of trovafloxacin while not affecting its main structure, sites 1- and 5- were modified in this study to replace CH
3 (site 1-) with five hydrophobic groups (-NO
2, -C
2H
5, -C
3H
7, -SH and -OCH
3) and H (site 5-) with two electronegative groups (-SiH
3 and -PH
2). Finally, a total of 16 TRO derivatives were produced (
Table 2).
3.3. Evaluation of Functionality, Environmental-Friendliness, and Stability of TRO Derivatives Based on HQSAR and QSAR Models
3.3.1. Functional Evaluation of TRO Derivatives Based on QSAR Model
The 3D-QSAR model of plasma protein binding rate constructed in this study and the HQSAR model of fluoroquinolone genotoxicity (pLOEC), both established by Zhao et al. [
14], were used to evaluate the functional characteristics (plasma protein binding rate and genotoxicity) of 16 TRO derivatives (
Table 2).
The plasma protein binding rates of 11 TRO derivatives decreased by different extents compared with that of trovafloxacin. The plasma protein binding rates of derivative-10 (1-C2H5-5-PH2-Trovafloxacin) and derivative-11 (1-C3H7-5-PH2-Trovafloxacin) decreased significantly by 24.18% and 20.60%, respectively. In addition, the genotoxicity (pLOEC) of the 11 TRO derivatives increased by different amounts compared to that of trovafloxacin (range of increase was 4.89–21.36%). Genotoxicity means that quinolones selectively inhibit two enzymes that play a role in DNA synthesis in bacteria, namely topoisomerases II and IV. This prevents replication, transcription, and repair of bacterial DNA, thus making it impossible for bacteria to grow and multiply. Therefore, increased genotoxicity can improve the medicinal effect of the drug.
In summary, the 11 TRO derivatives designed in this study have two beneficial effects on the human body, first they reduced the adverse effects (plasma protein binding rate), then they also enhanced the pharmacological effects (genotoxicity).
3.3.2. Evaluation of Environmental Friendliness of TRO Derivatives Based on 3D-QSAR Model
The QSAR model of bioconcentration (log
Kow) and photodegradation (log
t1/2) of fluoroquinolones constructed by Zhao et al. [
15,
16] was used to predict the bioconcentration and photodegradation of 11 TRO derivatives screened here (
Table 3).
The bioconcentration of 11 TRO derivatives decreased by 36.90–61.41%, while their photodegradability increased by 9.04–20.56%. The 11 TRO derivatives were less prone to enrichment than trovafloxacin in the environment, and the residual parts were more easily photodegraded.
3.3.3. Evaluation of Stability of TRO Derivatives Based on Density Functional Theory
The binding rate to the plasma protein of 11 TRO derivatives designed by using the CoMSIA model decreased to different amounts, and these decreases were beneficial from the viewpoint of human health. To further achieve the derivatization of fluoroquinolones, it is necessary to characterise the stability (stability in the environment) of the derivatives and to evaluate the difficulty of the reaction.
The positive frequency value of a molecule can directly reflect whether the molecule can remain stable in the environment. When the positive frequency value of a molecule is greater than zero, it can remain stable in the environment; otherwise, it cannot [
29]. Therefore, density functional theory was used to calculate the positive frequency values of 11 TRO derivatives to test their stability [
30] (
Table 4).
According to
Table 4, the positive frequency values of all the 11 TRO derivatives are greater than zero, so the 11 derivatives designed herein can remain stable in the environment.
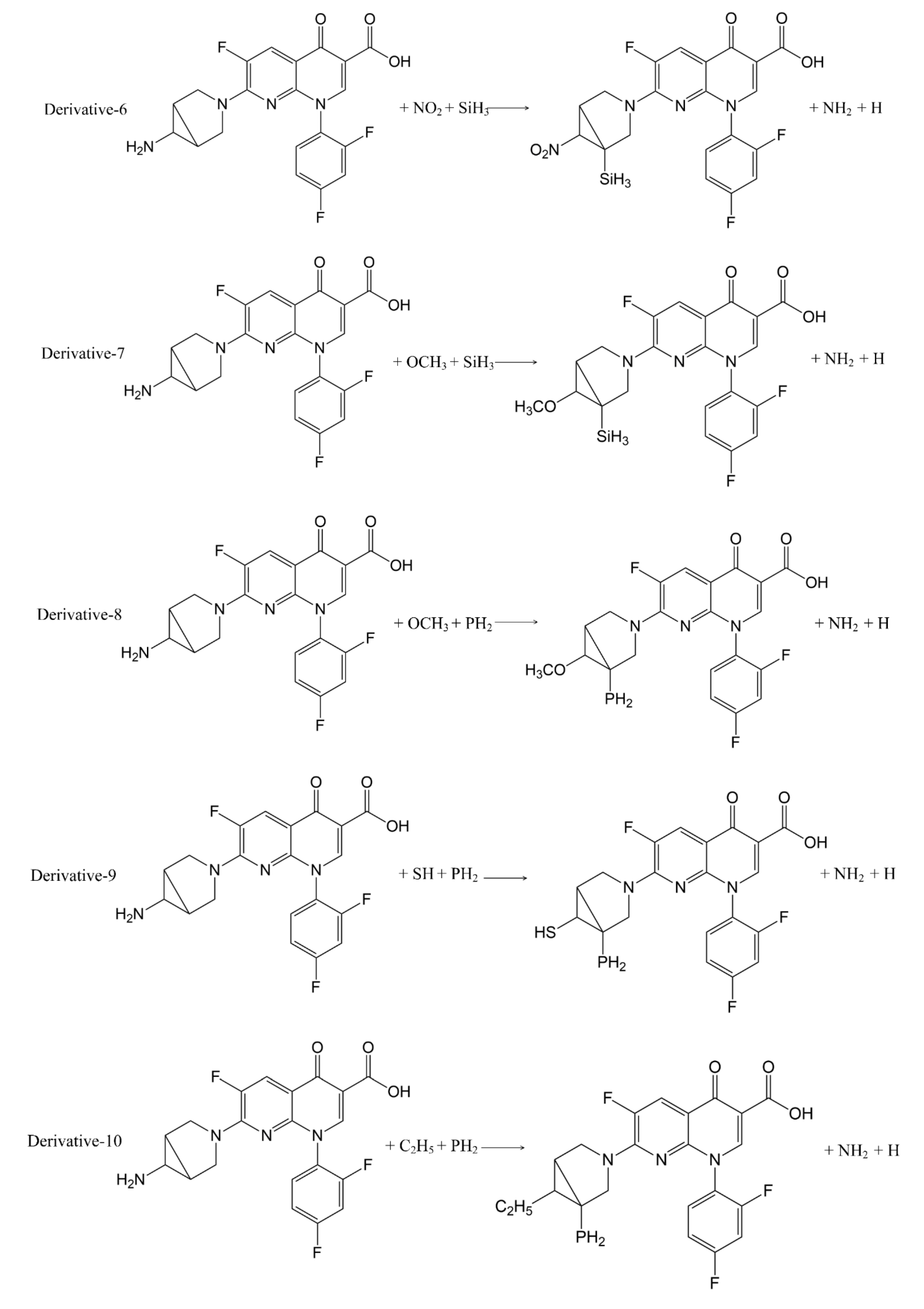
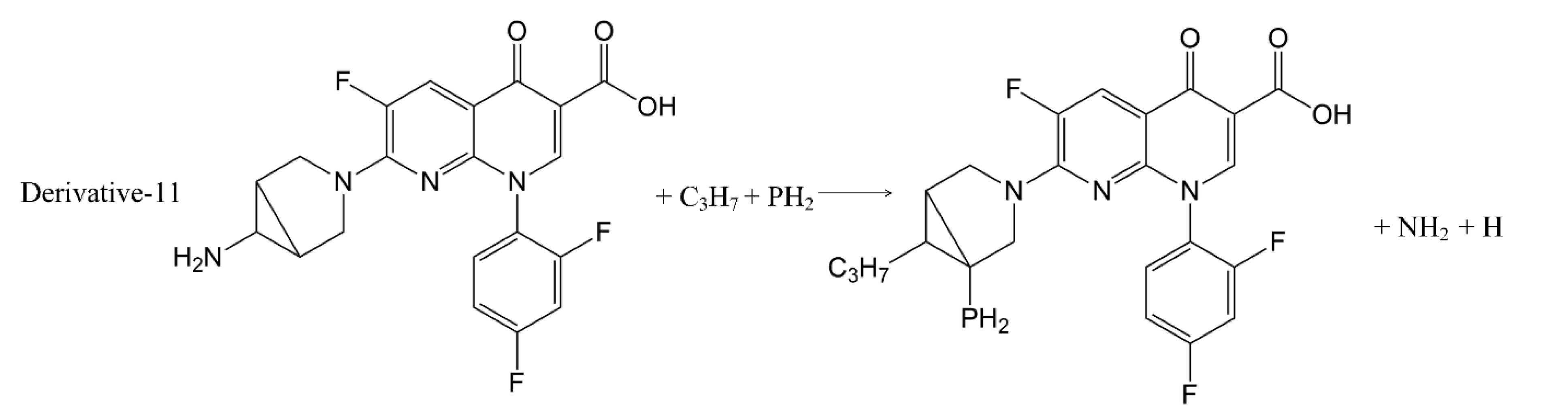
In addition to calculation of the positive frequency values of the TRO derivatives, the pathways of substitution reaction between the 7 substituents (-SH, -C
2H
5, -C
3H
7, -SiH
3, -NO
2, -PH
2, -OCH
3) and trovafloxacin were inferred (
Figure 3). The Gibbs free energy change (ΔG) of the 11 substitution reactions were calculated using Formula (2) to judge the possibility of occurrence of the substitution reaction pathways [
31,
32] (
Table 5).
The ΔG values of the 11 substitution reaction pathways were less than 0, indicating that the substitution reactions can proceed spontaneously, and that the inferred substitution reaction pathways were reasonable.
3.3.4. ADMET Prediction of Trovafloxacin Derivatives
DS software was used to predict the absorption (A), distribution (D), metabolism (M), excretion (E) and toxicity (T) of TRO and its derivatives [
33,
34,
35,
36,
37]. The aqueous solubility, human intestinal absorption, blood–brain barrier penetration, cytochrome P450 2D6 inhibition, hepatotoxicity and plasma protein binding rate were measured. Among them, the aqueous solubility of the molecule is closely related to the distribution and delivery of the drug in the body, and is one of the key factors for the preparation of a drug. Human intestinal absorption and blood–brain barrier can respectively influence the absorption of drugs from the intestines and enter the brain tissue to exert their drug effects. Cytochrome P450 is the main metabolic enzyme of drugs and other internal and external sources. Its activity may be inhibited or induced by drugs, which is essential for the metabolism of drugs in the body. In addition, the liver is an important organ for drug metabolism and is easily damaged by drugs; drugs will bind to plasma proteins at a certain ratio after being taken, so drugs are divided into bound and free types in the body, and only the free type has drug activity, that is, drugs are not combined with plasma protein [
38].
Good drugs should have suitable aqueous solubility, good blood–brain barrier penetration, low toxicity and no inhibition of cytochrome P450, and mainly present the free type in plasma and have good intestinal absorption [
39]. Through the prediction of TRO, derivative-10 and derivative-11, it was found that the 25 °C water solubility (the level is 2, low) and the human intestinal absorbability (the level is 0, good) of derivative-10 and derivative-11 relative to TRO were unchanged in the level. It showed that derivative-10 and derivative-11 have good solubility and intestinal absorption, and meet the standards of drug preparations. The blood–brain barrier penetration of the two derivatives was improved relative to TRO, hepatotoxicity was reduced, and there was no inhibitory effect on cytochrome P450 (Bayesian score was less than 0.162). However, the plasma protein binding rate of derivative-11 increased relative to TRO, while derivative-10 (level 0, good) remained unchanged. Therefore, among all derivatives, derivative-10 has the most characteristics of a new drug and is better than the target molecule TRO.
3.4. Analysis of Mechanism of Decrease in TRO Derivatives’ Plasma Protein Binding Rate Based on Molecular Docking
The target molecule and the 11 TRO derivatives were docked with plasma protein (ID:5NU7) by using Discovery Studio [
40], and the relationship between the scoring function and the binding rate was further discussed (
Table 6).
Except for derivative-1, the scores of the other fluoroquinolone derivatives were lower than that of trovafloxacin, and the level of decrease was consistent with the decrease in the plasma protein binding rate. For example, the plasma protein binding rates of derivative-10 and derivative-11 were 24.18% and 20.60% lower than that of trovafloxacin, respectively, and the corresponding docking results decreased by 24.43% and 17.83%. The above results further confirmed that the binding ability to plasma protein of the 11 TRO derivatives was reduced which compared with that of the target molecule.
3.4.1. Qualitative Analysis of Mechanism of Decrease in Plasma Protein Binding Rate of TRO Derivatives Based on Changes in Amino Acid Residue
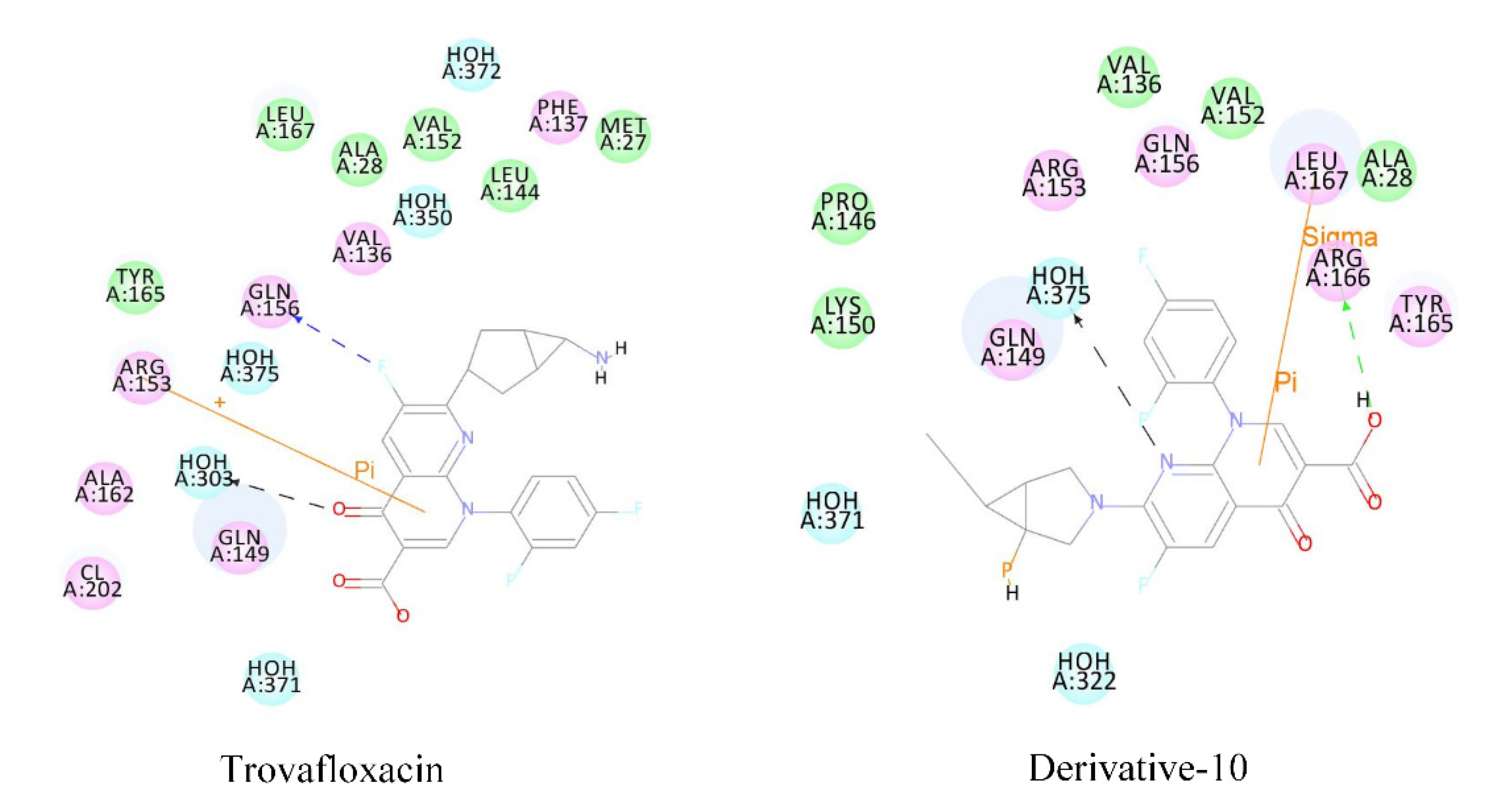
Fluoroquinolones as antigens can bind to plasma proteins and stimulate the body to produce drug antigen-specific antibodies, thus reducing the number of white blood cells. The plasma protein binding rate and docking score of derivative-10 decreased significantly. Therefore, we subjected molecular docking results of derivative 10 to mechanism analysis mainly by studying the polarity and number of amino acid residues that play major roles in the acceptor molecule when derivative-10 binds to plasma protein (
Table 7 and
Figure 4).
When trovafloxacin was docked with plasma protein, the amino acid residues making interactions could be divided into two categories; hydrophobic amino acids represented by Leu, Val, Phe, Met, Ala, Tyr, and hydrophilic amino acids represented by Gln and Arg. There were nine hydrophobic amino acid residues and three hydrophilic amino acid residues. When derivative-10 was docked with plasma protein, the hydrophobic amino acids were mainly Leu, Val, Pro, Ala, and Tyr, that is, a total of six amino acid residues, while the hydrophilic amino acid residues were mainly Gln, Lys, and Arg, that is, a total of five amino acid residues. This indicated that when quinolones enter into plasma protein composed of hydrophilic amino acids, the interaction between the receptor protein and the donor molecule weakens, and the decrease in the docking function score and the plasma protein binding rate are related to changes in amino acid residues.
3.4.2. Quantitative Analysis of the Decrease in Plasma Protein Binding Rate of TRO Derivatives
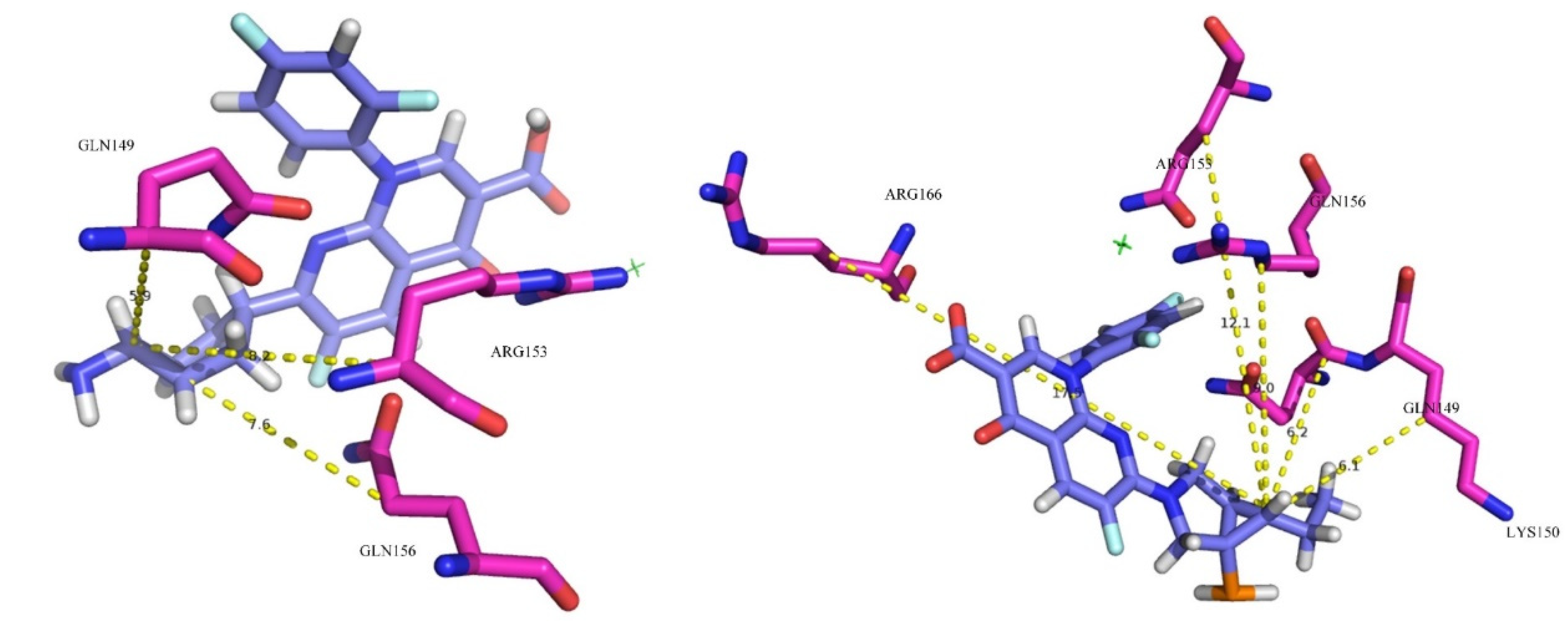
To explore the effect of hydrophilicity on the binding force between derivative-10 and plasma protein, the distances between substitution site 1- of Trovafloxacin and derivative-10 and the hydrophilic amino acid residues at the active site were measured and averaged (
Figure 5 and
Table 8).
The bond lengths between the NH
2 substituent at site 1 of trovafloxacin and the hydrophilic amino acid residues Gln149, Gln156, and Arg153 at the active site of the plasma protein were 5.9 Å, 7.6 Å, and 8.2 Å, respectively, and the average distance was 7.2 Å. The bond lengths between the C
2H
5 substituent at site 1 of derivative-10 and the hydrophilic amino acid residues Gln149, Gln156, Arg153, Arg166, and Lys150 at the active site of the plasma protein were 6.2 Å (>5.9 Å), 9.0 Å (>7.6Å), 12.1Å (>8.2 Å), 17.5 Å, and 6.1 Å, respectively, with an average distance of 10.2 Å. The binding rate and docking scoring function of trovafloxacin to plasma protein in
Table 2 and
Table 6 are higher than those of derivative-10. It was concluded that the further the average distance between the substituent of site 1- and the hydrophilic amino acid residue, the weaker is the hydrophilic effect between fluoroquinolone and plasma protein, and the smaller is the binding force between them. Therefore, the average distance between substituent 1- and the hydrophilic amino acid residue is negatively correlated with the binding force between fluoroquinolone and plasma protein, as well as with the plasma protein binding rate of fluoroquinolone.
The further the distance (average distance) between substitution site 1- of derivative-10 and the hydrophilic amino acid residues at the plasma protein binding site is, the more difficult it is to form a strong hydrophilic interaction between fluoroquinolone and plasma protein. Therefore, the weaker the binding force between fluoroquinolone and plasma protein, the lower is the plasma protein binding rate of fluoroquinolone. Thus, this study further explains the reason for the difference in binding between TRO derivatives and plasma protein.
3.5. Interaction Analysis between TRO Derivative and Plasma Protein Based on Molecular Dynamics Simulation
Molecular dynamics simulations [
41,
42] mainly employ the basic principles of Newtonian mechanics, taking molecular motion as the main simulation object and studying the dynamic process of the motion state of all particles in the system with time. Molecular dynamics simulations can provide detailed information about changes in protein structure with time. Moreover, the calculation results of molecular dynamics simulations are very similar to real experimental results in many aspects, and they can be used to test the rationality of statistical mechanics theory and to study the effect of microscopic forces on macroscopic properties. Since Mccammon et al. [
43] first used molecular dynamics simulations to study the dynamic properties of bovine insulin inhibitors in 1977, this method has been used to study drugs, protein molecules, and in other fields [
44,
45,
46].
3.5.1. Analysis of Molecular Trajectories of TRO Derivatives Based on RMSD and RMSF
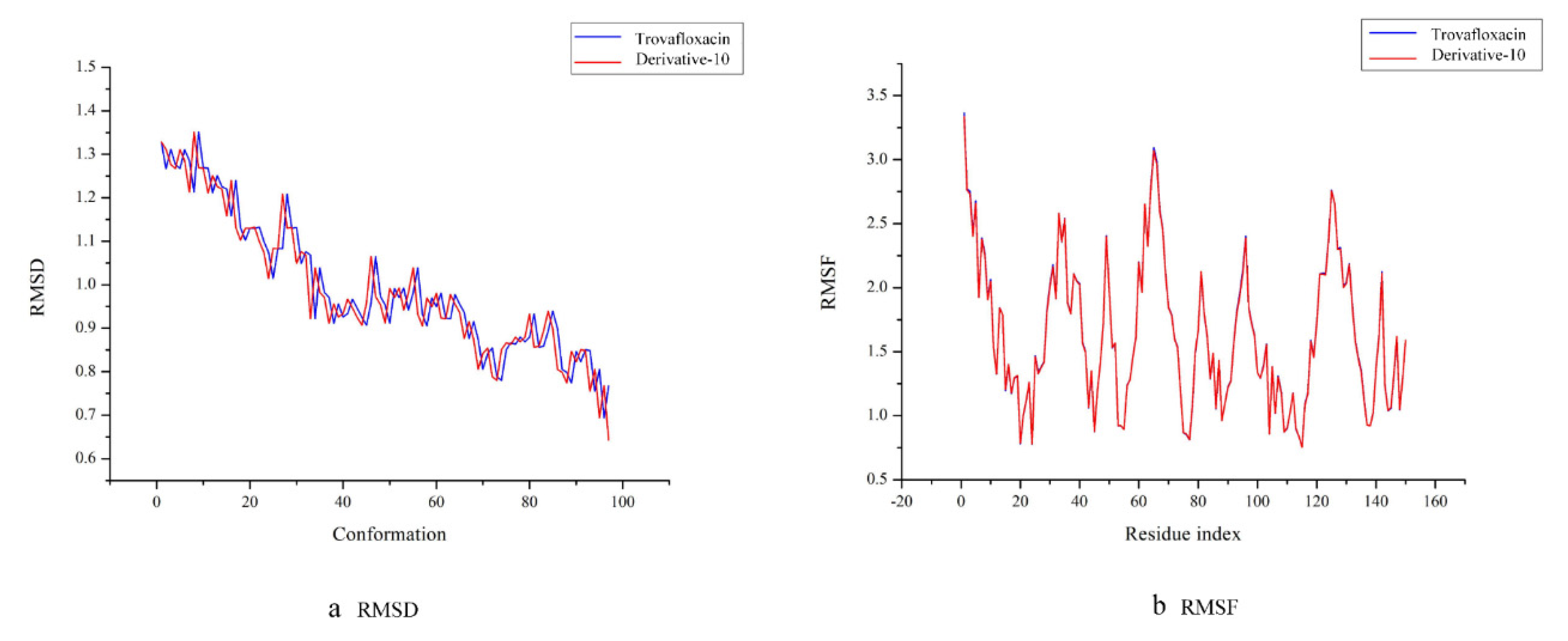
Molecular dynamics simulations of the complexes of trovafloxacin and derivative-10 with plasma protein were performed, and their trajectories were analysed based on root mean square deviation (RMSD) and root mean square fluctuation (RMSF).
RMSD refers to the root mean square deviation of each atom between the protein conformation and its initial conformation. Each conformation has an RMSD value. The smaller the RMSD value is, the lighter is the motion amplitude of each atom in the protein and the higher is its stability [
47].
Figure 6a shows that the RMSD trajectories of trovafloxacin and derivative-10 are essentially identical, which means that during molecular dynamics simulation, trovafloxacin and derivative-10 can be thought to have exhibited good stability. In addition, in the first 40 conformations, the motion trajectory showed a significant downward trend, and as a result, the trajectories of the complexes fluctuate greatly at this stage. When the simulation system reached the 40th conformation, the RMSD value of the entire system was low, and the fluctuation amplitude was small. As a result, all systems tended to be balanced at this time.
The RMSF value refers to the root mean square fluctuation of amino acid residues, which reflects the conformation of each amino acid residue in the molecular dynamic trajectory. A high RMSF value indicates a less stable conformation of amino acids and vice versa [
48]. According to
Figure 6b, the RMSF trajectories of trovafloxacin and derivative-10 with the plasma protein complexes are essentially identical. With a small number of amino acid residues, the RMSF value of trovafloxacin was slightly higher than that of derivative-10. Therefore, compared to trovafloxacin, derivative-10 can be considered more stable. Moreover, the amino acid residues of trovafloxacin and derivative-10 fluctuated widely in the initial stage, which might be caused by the fact that the interaction area at this stage was an unstable region of plasma protein. Then, exhibited a staged decline trend wax, indicating that the entire system tended to stable. The lowest fluctuation of amino acid residues occurred in the range of 100–120, indicating that the stability of the complexes was the highest in this stage.
3.5.2. Qualitative and Semi-quantitative Analysis of the Molecular Trajectories Based on Potential Energy and Total Energy
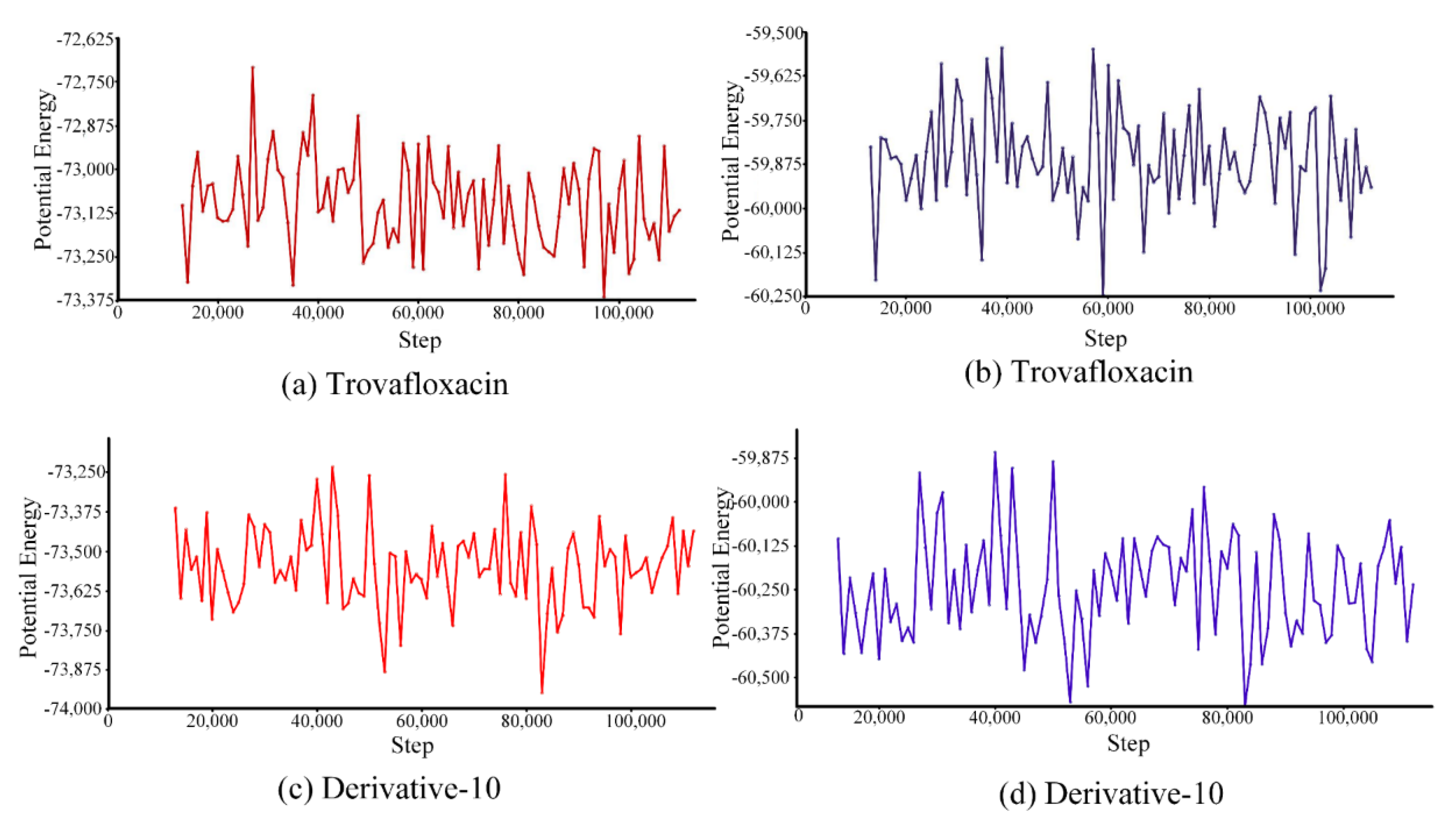
Energy simulation was performed for the complexes of trovafloxacin and derivative-10 with plasma protein (
Figure 7).
Because of the interaction force between molecules, energy is related to their relative positions, namely molecular potential energy and total energy [
49]. The motion trajectory of potential energy (
Figure 7a,c) showed that the potential energy of trovafloxacin varied between −73,375 kcal/mol and −72,625 kcal/mol, and its average potential energy was approximately −73,100 kcal/mol. However, the potential energy of derivative-10 ranged from −73,875 kcal/mol to −73,250 kcal/mol, and the average potential energy was approximately −73,500 kcal/mol. Therefore, the range of potential energy and average potential energy of the combination of derivative-10 and plasma protein were lower than those of trovafloxacin combined with plasma protein. This result indicates that the interaction between derivative-10 and plasma proteins was weaker.
In addition to studying changes in the potential energy of trovafloxacin and derivative-10, we further analysed the total energy in the simulated system (
Figure 7b,d). The total energy of trovafloxacin combined with plasma protein varied between −60,250 kcal/mol and −59,500 kcal/mol, and the average total energy was approximately −59,875 kcal/mol. The total energy change of derivative-10 after binding to plasma protein ranged from −60,500 kcal/mol to −59,875 kcal/mol, with an average total energy of approximately −60,250 kcal/mol. Therefore, the total energy variation range and the average total energy of derivative-10 were lower than those of trovafloxacin.
The interaction energy between derivative-10 and plasma protein was lower than that between trovafloxacin and plasma protein in the molecular dynamics simulation system, further explained the decrease in the binding rate of trovafloxacin derivatives to plasma proteins.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}