
Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing

 , , , , , , , , and
, , , , , , , , and 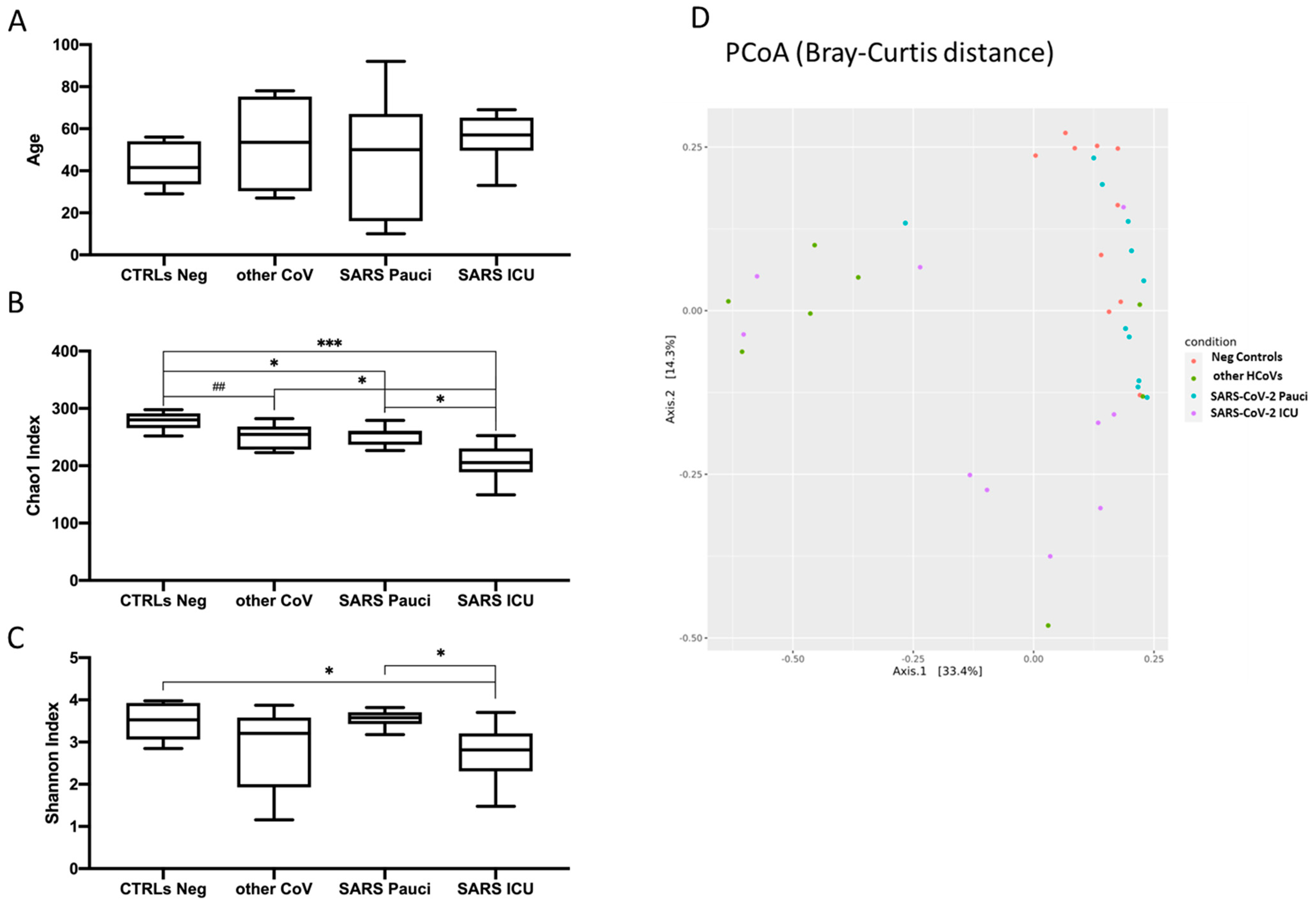{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Virus Detection
2.3. Sample Collection, DNA Extraction and Next-Generation Sequencing of Bacterial 16S Ribosomal RNA Gene
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
3.1. Study Population
3.2. Microbial Richness and Diversity Indices in SARS-CoV-2 and Other HCoVs Patients as Compared to Neg Controls
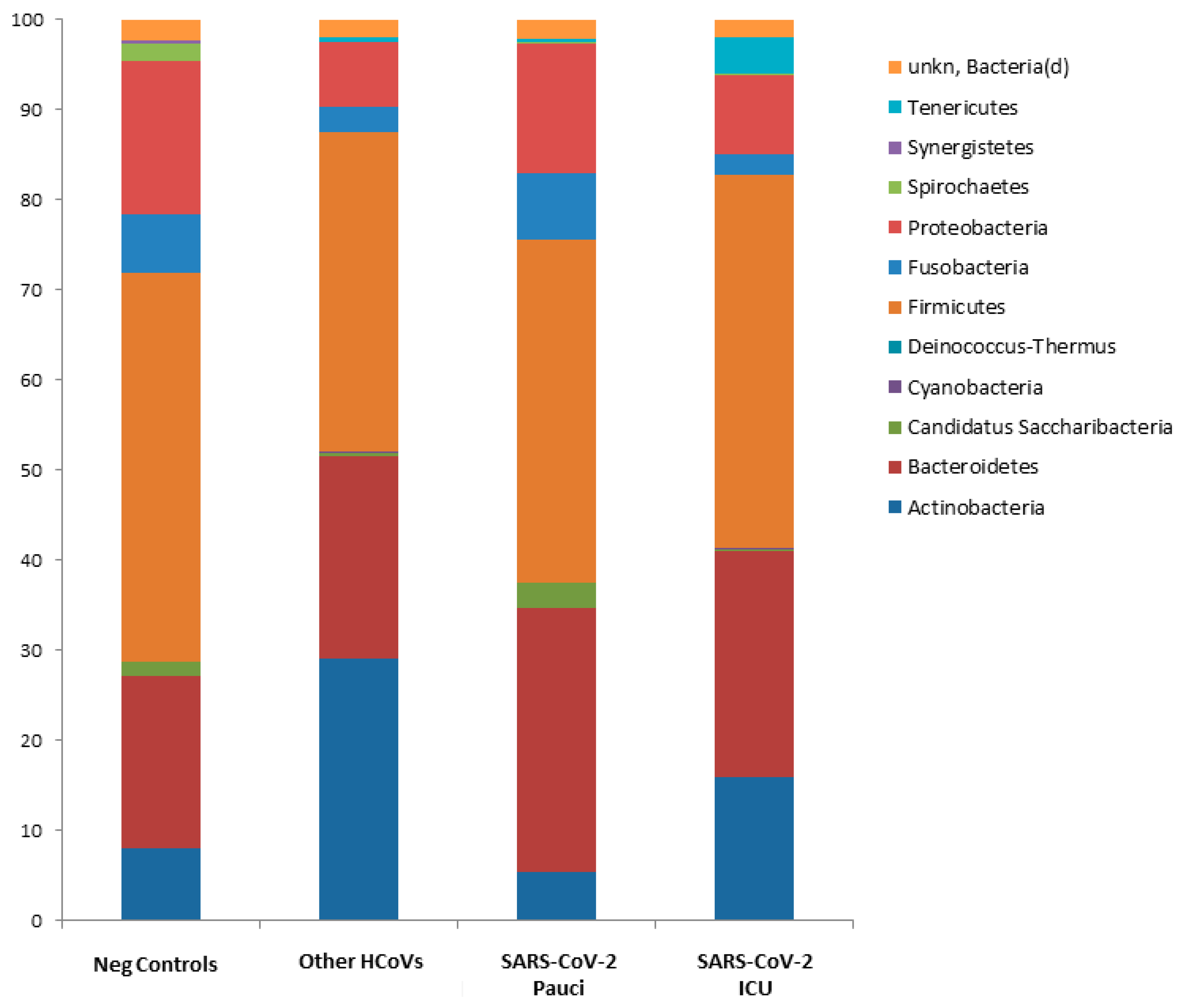
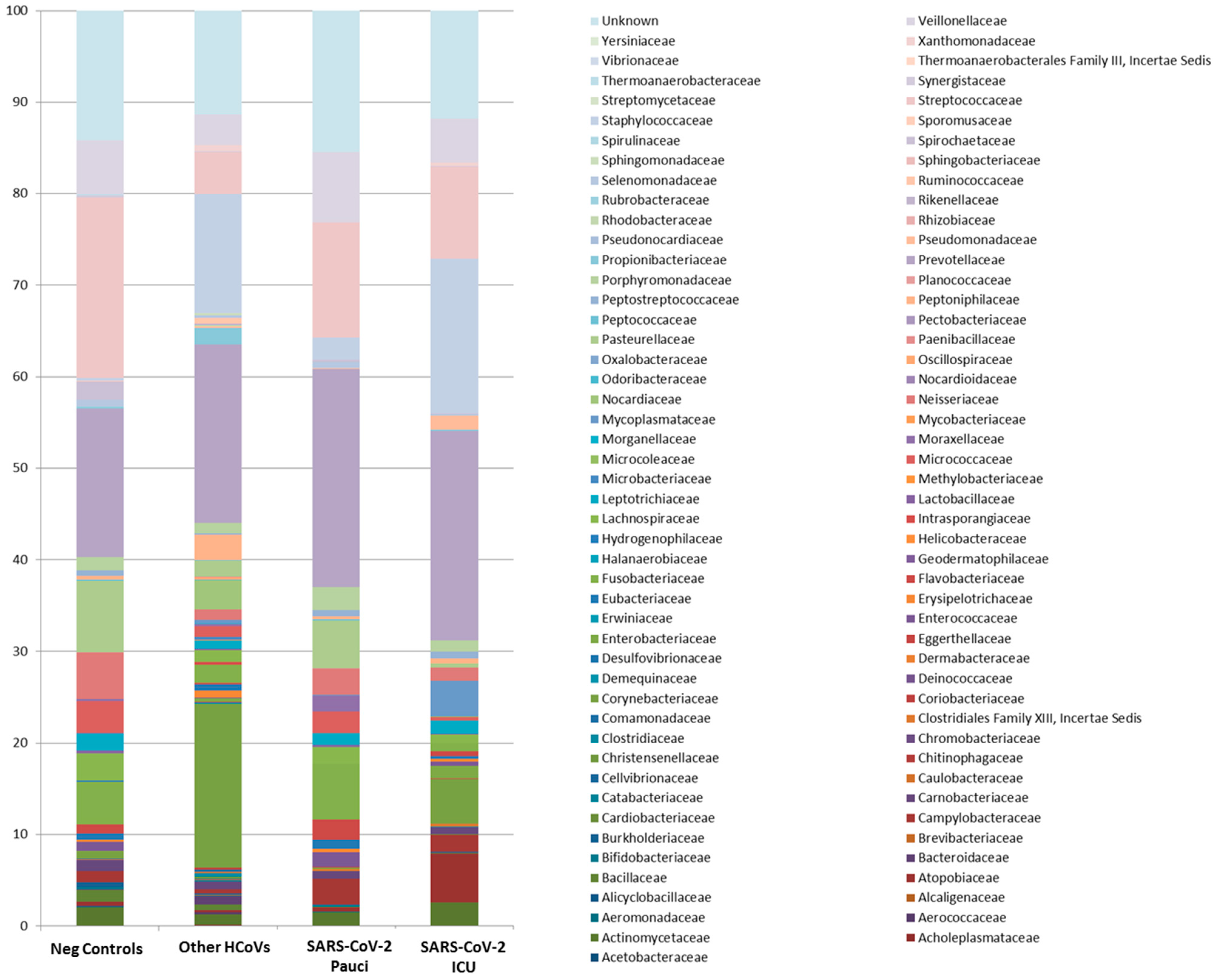
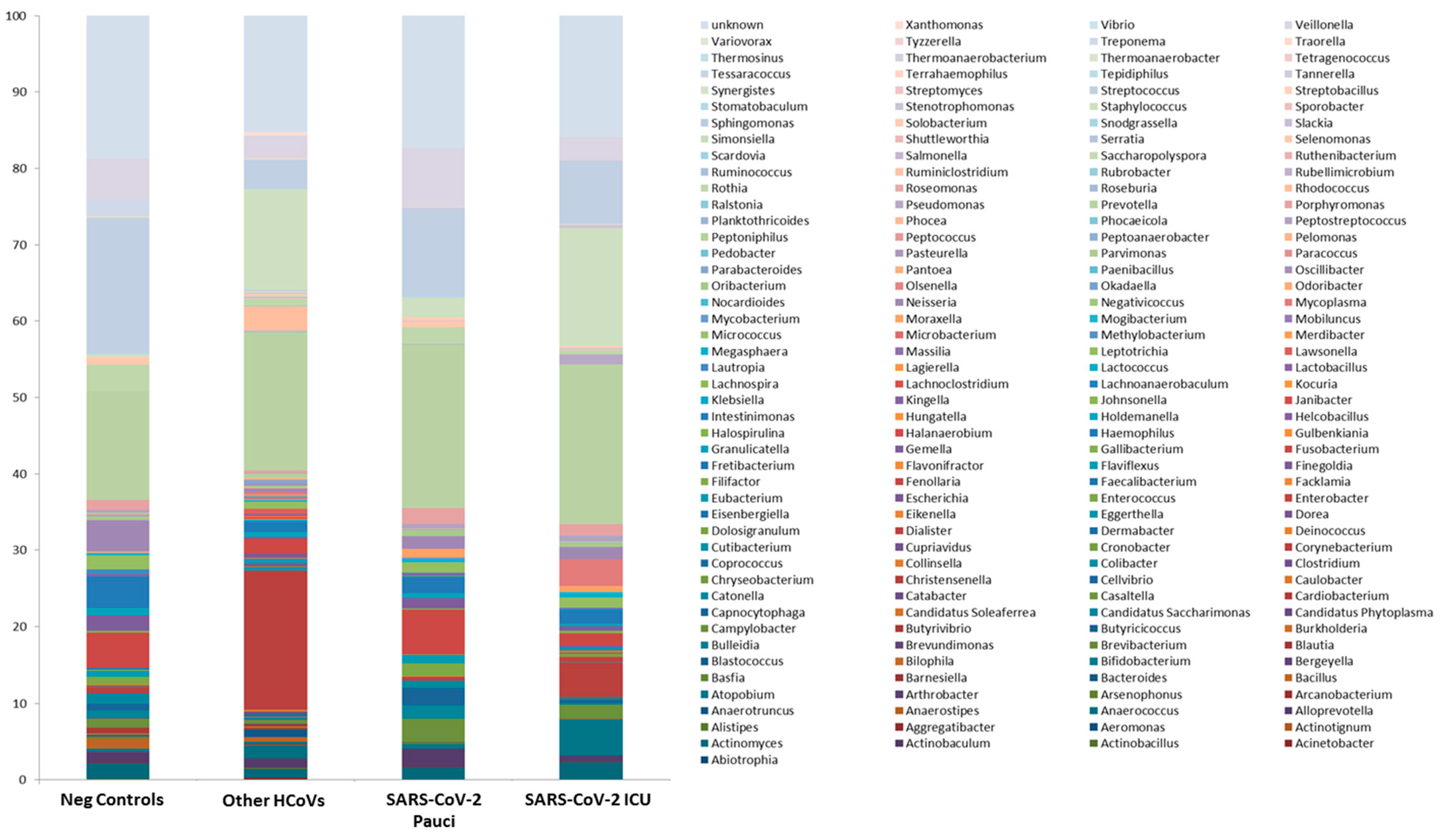
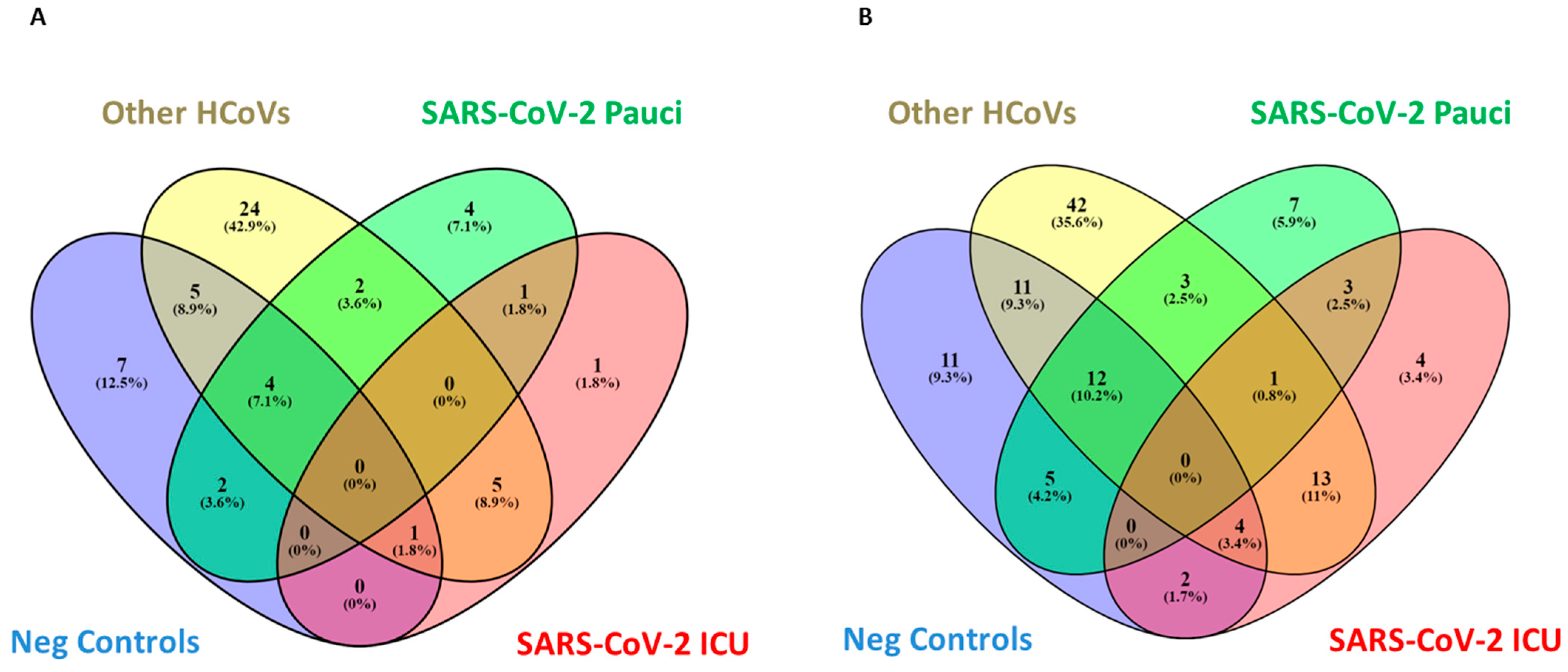
3.3. Nasal/Oropharyngeal Microbiota in SARS-CoV-2 Patients and Other HCoVs as Compared to Controls
3.4. Nasal/Ooropharyngeal Microbiota of COVID-19 Patients in Intensive Care Unit as Compared to COVID-19 Paucisymptomatic Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Bart, L.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; et al. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Liu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.W.; Wu, X.X.; Jiang, X.G.; Xu, K.J.; Ying, L.J.; Ma, C.L.; Li, S.-B.; Wang, H.-Y.; Zhang, S.; Gao, H.-N.; et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: Retrospective case series. BMJ 2020, 368, m606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zeng, W.; Li, X.; Chen, H.; Shi, L.; Li, X.; Xiang, H.; Cao, Y.; Chen, H.; Liu, C.; et al. CT imaging changes of corona virus disease 2019(COVID-19): A multi-center study in Southwest China. J. Transl. Med. 2020, 18, 154. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Zhang, X.; Qu, J. Coronavirus disease 2019 (COVID-19): A clinical update. Front. Med. 2020, 14, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Misra, A. Contentious issues and evolving concepts in the clinical presentation and management of patients with COVID-19 infection with reference to use of therapeutic and other drugs used in Co-morbid diseases (Hypertension, diabetes etc). Diabetes Metab. Syndr. 2020, 14, 251–254. [Google Scholar] [CrossRef]
- Wilks, J.; Golovkina, T. Influence of microbiota on viral infections. PLoS Pathog. 2012, 5, e1002681. [Google Scholar] [CrossRef] [Green Version]
- Idris, A.; Hasnain, S.Z.; Huat, L.U.Z.; Koh, D. Human diseases, immunity and the oral microbiota—Insights gained from metagenomic studies. Oral Sci. Int. 2017, 14, 27–32. [Google Scholar] [CrossRef]
- Di Stadio, A.; Costantini, C.; Renga, G.; Pariano, M.; Ricci, G.; Romani, L. The Microbiota/Host Immune System Interaction in the Nose to Protect from COVID-19. Life 2020, 10, 345. [Google Scholar] [CrossRef]
- De Steenhuijsen Piters, W.A.A.; Heinonen, S.; Hasrat, R.; Bunsow, E.; Smith, B.; Suarez-Arrabal, M.-C.; Chaussabel, D.; Cohen, D.M.; Sanders, E.A.M.; Ramilo, O.; et al. Naso-pharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 2016, 194, 1104–1115. [Google Scholar] [CrossRef]
- Harata, G.; He, F.; Hiruta, N.; Kawase, M.; Kubota, A.; Hiramatsu, M.; Yausi, H. Intranasal administration of Lactobacillus rhamnosus GG protects mice from H1N1 influenza vi rusinfection by regulating respiratory immune responses. Lett. Appl. Microbiol. 2010, 50, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.A.; Kainz, K.; Carmona-Gutierrez, D.; Madeo, F. Microbial wars: Competition in ecological niches and within the microbiome. Microb. Cell 2018, 5, 215–219. [Google Scholar] [CrossRef]
- Panebianco, C.; Latiano, T.; Pazienza, V. Microbiota Manipulation by Probiotics Administration as Emerging Tool in Cancer Prevention and Therapy. Front. Oncol. 2020, 10, 679. [Google Scholar] [CrossRef]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the drug-microbiota interactions in anticancer therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Fontana, A.; Panebianco, C.; Picchianti-Diamanti, A.; Laganà, B.; Cavalieri, D.; Potenza, A.; Pracella, R.; Binda, E.; Copetti, M.; Pazienza, V. Gut Microbiota Profiles Differ among Individuals Depending on Their Region of Origin: An Italian Pilot Study. Int. J. Environ. Res. Public Health 2019, 16, 4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanaspa, M.; Bassat, Q.; Medeiros, M.M.; Muñoz-Almagro, C. Respiratory microbiota and lower respiratory tract disease. Expert Rev. Anti. Infect Ther. 2017, 15, 703–711. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dos Anjos Borges, L.G.; Giongo, A.; de Mattos Pereira, L.; Trindade, F.J.; Gregianini, T.S.; Campos, F.S.; Ghedin, E.; Gorini da Veiga, A.B. Comparison of the nasopharynx microbiome between influenza and non-influenza cases of severe acute respiratory infections: A pilot study. Health Sci. Rep. 2018, 1, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rueca, M.; Fontana, A.; Bartolini, B.; Piselli, P.; Mazzarelli, A.; Copetti, M.; Binda, E.; Perri, F.; Gruber, C.E.M.; Nicastri, E.; et al. Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing. Int. J. Environ. Res. Public Health 2021, 18, 2174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18042174
Rueca M, Fontana A, Bartolini B, Piselli P, Mazzarelli A, Copetti M, Binda E, Perri F, Gruber CEM, Nicastri E, et al. Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing. International Journal of Environmental Research and Public Health. 2021; 18(4):2174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18042174
Chicago/Turabian StyleRueca, Martina, Andrea Fontana, Barbara Bartolini, Pierluca Piselli, Antonio Mazzarelli, Massimiliano Copetti, Elena Binda, Francesco Perri, Cesare Ernesto Maria Gruber, Emanuele Nicastri, and et al. 2021. "Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing" International Journal of Environmental Research and Public Health 18, no. 4: 2174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18042174