Central Apneas Due to the CLIFAHDD Syndrome Successfully Treated with Pyridostigmine

 , , ,
, , ,
Abstract
:1. Introduction
2. Case Description
3. Discussion
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gao, S.; Xie, L.; Kawano, T.; Po, M.D.; Pirri, J.K.; Guan, S.; Alkema, M.J.; Zhen, M. The NCA sodium leak channel is required for persistent motor circuit activity that sustains locomotion. Nat. Commun. 2015, 6, 6323. [Google Scholar] [CrossRef] [PubMed]
- Al-Sayed, M.D.; Al-Zaidan, H.; Albakheet, A.; Hakami, H.; Kenana, R.; Al-Yafee, Y.; Al-Dosary, M.; Qari, A.; Al-Sheddi, T.; Al-Muheiza, M.; et al. Mutations in NALCN Cause an Autosomal-Recessive Syndrome with Severe Hypotonia, Speech Impairment, and Cognitive Delay. Am. J. Hum. Genet. 2013, 93, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; McMillin, M.J.; Shively, K.M.; Beck, A.E.; Marvin, C.T.; Armenteros, J.R.; Buckingham, K.J.; Nkinsi, N.T.; Boyle, E.; Berry, M.N.; et al. De Novo Mutations in NALCN Cause a Syndrome Characterized by Congenital Contractures of the Limbs and Face, Hypotonia, and Developmental Delay. Am. J. Hum. Genet. 2015, 96, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, L.; Shen, D.; Ding, C.; Yang, X.; Zhang, W.; Li, J.; Deng, J.; Gong, S.; Liu, J.; et al. Compound Heterozygous CHAT Gene Mutations of a Large Deletion and a Missense Variant in a Chinese Patient With Severe Congenital Myasthenic Syndrome With Episodic Apnea. Front. Pharmacol. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- McMacken, G.; Whittaker, R.; Evangelista, T.; Abicht, A.; Dusl, M.; Lochmüller, H. Congenital myasthenic syndrome with episodic apnoea: Clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J. Neurol. 2017, 265, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.C.; Morrison, D.E.; Ellenberger, H.H.; Otto, M.R.; Feldman, J.L. Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J. Comp. Neurol. 1989, 281, 69–96. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, P.G.; Bayliss, D.A. Neural Control of Breathing and CO2 Homeostasis. Neuron 2015, 87, 946–961. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.L.; Del Negro, C.A.; Gray, P.A. Understanding the Rhythm of Breathing: So Near, Yet So Far. Annu. Rev. Physiol. 2013, 75, 423–452. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.M.; Baertsch, N.A.; Moreira, T.S.; Ramirez, J.-M.; Takakura, A.C. Unraveling the Mechanisms Underlying Irregularities in Inspiratory Rhythm Generation in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2021, 41, 4732–4747. [Google Scholar] [CrossRef]
- Atherton, J.F.; Bevan, M.D. Ionic Mechanisms Underlying Autonomous Action Potential Generation in the Somata and Dendrites of GABAergic Substantia Nigra Pars Reticulata Neurons In Vitro. J. Neurosci. 2005, 25, 8272–8281. [Google Scholar] [CrossRef] [Green Version]
- Kschonsak, M.; Chua, H.C.; Noland, C.L.; Weidling, C.; Clairfeuille, T.; Bahlke, O.; Ameen, A.O.; Li, Z.R.; Arthur, C.P.; Ciferri, C.; et al. Structure of the human sodium leak channel NALCN. Nature 2020, 587, 313–318. [Google Scholar] [CrossRef]
- Lu, B.; Su, Y.; Das, S.; Liu, J.; Xia, J.; Ren, D. The Neuronal Channel NALCN Contributes Resting Sodium Permeability and Is Required for Normal Respiratory Rhythm. Cell 2007, 129, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Abe, C.; Holloway, B.B.; Shu, S.; Kumar, N.; Weaver, J.L.; Sen, J.; Perez-Reyes, E.; Stornetta, R.L.; Guyenet, P.G.; et al. NALCN Is a “Leak” Sodium Channel That Regulates Excitability of Brainstem Chemosensory Neurons and Breathing. J. Neurosci. 2016, 36, 8174–8187. [Google Scholar] [CrossRef]
- Xie, J.; Ke, M.; Xu, L.; Lin, S.; Huang, J.; Zhang, J.; Yang, F.; Wu, J.; Yan, Z. Structure of the human sodium leak channel NALCN in complex with FAM155A. Nat. Commun. 2020, 11, 5831. [Google Scholar] [CrossRef]
- Maggi, L.; Mantegazza, R.; Mantegazza, R. Treatment of Myasthenia Gravis: Focus on pyridostigmine. Clin. Drug Investig. 2011, 31, 691–701. [Google Scholar] [CrossRef]
- Amourette, C.; Lamproglou, I.; Barbier, L.; Fauquette, W.; Zoppe, A.; Viret, R.; Diserbo, M. Gulf War illness: Effects of repeated stress and pyridostigmine treatment on blood–brain barrier permeability and cholinesterase activity in rat brain. Behav. Brain Res. 2009, 203, 207–214. [Google Scholar] [CrossRef]
- Friedman, A.; Kaufer, D.; Shemer, J.; Hendler, I.; Soreq, H.; Tur-Kaspa, I. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med. 1996, 2, 1382–1385. [Google Scholar] [CrossRef]
- Song, X.; Tian, H.; Bressler, J.; Pruett, S.; Pope, C. Acute and Repeated Restraint Stress Have Little Effect on Pyridostigmine Toxicity or Brain Regional Cholinesterase Inhibition in Rats. Toxicol. Sci. 2002, 69, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Grauer, E.; Alkalai, D.; Kapon, J.; Cohen, G.; Raveh, L. Stress Does Not Enable Pyridostigmine to Inhibit Brain Cholinesterase after Parenteral Administration. Toxicol. Appl. Pharmacol. 2000, 164, 301–304. [Google Scholar] [CrossRef]
- Kaufer, D.; Friedman, A.; Seidman, S.; Soreq, H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 1998, 393, 373–377. [Google Scholar] [CrossRef]
- Pavlovsky, L.; Browne, R.; Friedman, A. Pyridostigmine enhances glutamatergic transmission in hippocampal CA1 neurons. Exp. Neurol. 2003, 179, 181–187. [Google Scholar] [CrossRef]
- Pavlovsky, L.; Bitan, Y.; Shalev, H.; Serlin, Y.; Friedman, A. Stress-induced altered cholinergic–glutamatergic interactions in the mouse hippocampus. Brain Res. 2012, 1472, 99–106. [Google Scholar] [CrossRef]
- Lockhart, B.; Closier, M.; Howard, K.; Steward, C.; Lestage, P. Differential inhibition of [3H]-oxotremorine-M and [3H]-quinuclinidyl benzilate binding to muscarinic receptors in rat brain membranes with acetylcholinesterase inhibitors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 363, 429–438. [Google Scholar] [CrossRef]
- Swayne, L.A.; Mezghrani, A.; Varrault, A.; Chemin, J.; Bertrand, G.; Dalle, S.; Bourinet, E.; Lory, P.; Miller, R.J.; Nargeot, J.; et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic β-cell line. EMBO Rep. 2009, 10, 873–880. [Google Scholar] [CrossRef]
- Fukai, R.; Saitsu, H.; Okamoto, N.; Sakai, Y.; Fattal-Valevski, A.; Masaaki, S.; Kitai, Y.; Torio, M.; Kojima-Ishii, K.; Ihara, K.; et al. De novo missense mutations in NALCN cause developmental and intellectual impairment with hypotonia. J. Hum. Genet. 2016, 61, 451–455. [Google Scholar] [CrossRef]


{kind=link}
| Pregnancy | 2nd Pregnancy, 2nd Delivery in 36 Hbd. Birth Weight 2920 g, Apgar 10. Polyhydramnios (Third Trimester), Diabetes Mellitus. |
|---|---|
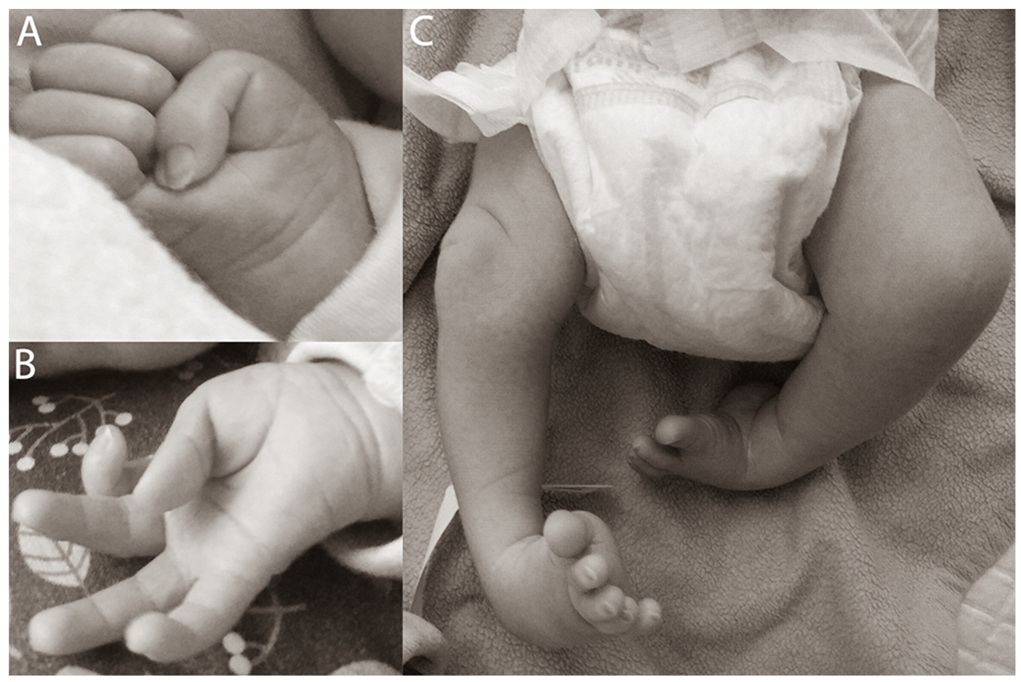
| Perinatal period | Clubfoot (manual redresion, plaster casts were used). Adducted thumbs. |
| MRI of head | 1st MRI (perinatal period)—normal. 2nd MRI (12 month)—slight enlargement of ventricular and subarachnoid space. |
| EMG | Failed to perform a full study. Routine examination showed no abnormalities. |
| EEG | 1st EEG (perinatal period) and 2nd EEG (12 month)—in normal range. |
| Laboratory tests | GC/MS profiling of urinary organic acids, cerebrospinal fluid, test for congenital metabolic defects performed by tandem mass spectroscopy (perinatal)—normal. |
| Cardiological examination | ECG, ECHO—normal. |
| Pulmonary examination | Chest X-ray, bronchofiberoscopy—normal. |
| Gastroenterological examination | Gastroesophageal reflux |
| Orthopedic examination | Distal arthrogryposis |
| Dysmorphologic examination | Adducted thumbs, pits over large joints (knees, elbows), equinovarus feet, hypotonic face, without dysmorphic features. |
| Neurological examination | At birth: normal. At 2 weeks of age: No control of the head position in the traction test, no spontaneous control of the head position, no grasping of objects with hands. However, eye contact with the patient and cognitive development were normal. At 7.5 months of age: Psychomotor delay, no speech, the patient was lying down and he could turn sideways on his own, started to grasp objects, no control of the head position. Decreased muscle tone in the long axis with correct muscle tone in limbs. At 12 months of age: Alternating divergent strabismus present. Muscle tonus was still reduced within the trunk. Tendon reflexes were symmetrical, no neurological pathological signs were found. As before, there was no spontaneous control of the head position and during the traction test. In the horizontal position, spontaneous limb movements with the correct pattern were present. His thumbs were adducted. The patient was still lying down, and he could only turn sideways on his own. He maintained normal eye contact. After starting pyridostigmine treatment: Spontaneous improvement in head position control and in traction test. Patient began to crawl and sit alone. |
| Apnea episodes | At the age of 2 weeks: First episodes of skin graying, decreased muscle tension and rolling back of eyes. At the age of 3 months: The frequency of episodes increased. Initially, episodes began with a decreased muscle tone, which subsequently increased. Loss of consciousness was also present. Episodes lasted up to one minute. At the age of 12 months: Symptoms worsened. Episodes lasted over 3 min, with loss of consciousness and defecation. Choking has been observed several times. Episodes were provoked by crying or medical procedures. At night, O2 saturation drops to 70 percent without accompanying clinical symptoms. After starting pyridostigmine: Reduction in the number of apnea attacks. There were no episodes requiring urgent hospitalization or respiratory resuscitation. Due to the nocturnal O2 saturation reduction, overnight CPAP was recommended. |
| Genetic examination | Karyotype: 46, XY. Array-CGH—normal. Sanger sequencing of the entire coding sequence of the ELP1 gene—normal. MLPA od the SMN1 gene—normal. The single WES study (Cegat, Tübingen, Germany) showed one possibly pathogenic variant: c.1870G > C, (p.Glu603Gln) in the NALCN gene. Further Sanger sequencing in the patient‘s parents indicated that the variant aroused de novo. |
| Clinical Features | Patients Described by Chong et al. [3] | Patient Reported by Us |
|---|---|---|
| Downslanting palpebral fissures | 10/14 | − |
| Strabismus | 7/14 | + |
| Esotropia | 3/714 | + |
| Broad nasal bridge | 14/14 | − |
| Anterverted nasal tip | 12/14 | − |
| Large nares | 14/14 | − |
| Short columella | 14/14 | − |
| Long philtrum | 12/14 | − |
| Micrognathia | 13/14 | − |
| Pursed lips | 9/14 | − |
| H-shaped dimpled chin | 8/14 | − |
| Deep nasolabial folds | 12/14 | − |
| Full cheeks | 13/14 | − |
| Camptodactyly | 14/14 | + |
| Ulnar deviation | 14/14 | − |
| Adducted thumbs | 14/14 | + |
| Calcaneovalgus deformity | 8/14 | + |
| Club foot | 8/14 | + |
| Hip contractures | 11/14 | + |
| Elbow contractures | 7/14 | − |
| Knee contractures | 9/14 | − |
| Scoliosis | 6/14 | − |
| Short neck | 10/14 | + |
| Cognitive delay | 11/14 | + |
| Speech delay | 12/14 | + |
| Motor delay | 14/14 | + |
| Seizures | 2/14 | − |
| Hypotonia | 7/14 | + |
| Respiratory insufficiency | 8/14 | + |
| Abnormal respiratory pattern in newborn period | 9/14 | − |
| Excessive drooling | 4/14 | + |
| Gastroesophageal reflux disease (GERD) | 9/14 | + |
| Constipation | 5/14 | − |
| Hernia | 9/14 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winczewska-Wiktor, A.; Hirschfeld, A.S.; Badura-Stronka, M.; Wojsyk-Banaszak, I.; Sobkowiak, P.; Bartkowska-Śniatkowska, A.; Babak, V.; Steinborn, B. Central Apneas Due to the CLIFAHDD Syndrome Successfully Treated with Pyridostigmine. Int. J. Environ. Res. Public Health 2022, 19, 775. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph19020775
Winczewska-Wiktor A, Hirschfeld AS, Badura-Stronka M, Wojsyk-Banaszak I, Sobkowiak P, Bartkowska-Śniatkowska A, Babak V, Steinborn B. Central Apneas Due to the CLIFAHDD Syndrome Successfully Treated with Pyridostigmine. International Journal of Environmental Research and Public Health. 2022; 19(2):775. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph19020775
Chicago/Turabian StyleWinczewska-Wiktor, Anna, Adam Sebastian Hirschfeld, Magdalena Badura-Stronka, Irena Wojsyk-Banaszak, Paulina Sobkowiak, Alicja Bartkowska-Śniatkowska, Valeriia Babak, and Barbara Steinborn. 2022. "Central Apneas Due to the CLIFAHDD Syndrome Successfully Treated with Pyridostigmine" International Journal of Environmental Research and Public Health 19, no. 2: 775. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph19020775