
Prognosis and Survival in Idiopathic Pulmonary Fibrosis in the Era of Antifibrotic Therapy in Italy: Evidence from a Longitudinal Population Study Based on Healthcare Utilization Databases
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population, Data Sources, Incident IPF Case Definition
2.2. Variables
2.3. Outcomes
2.4. Statistical Analysis
3. Results
3.1. In-Hospital Mortality
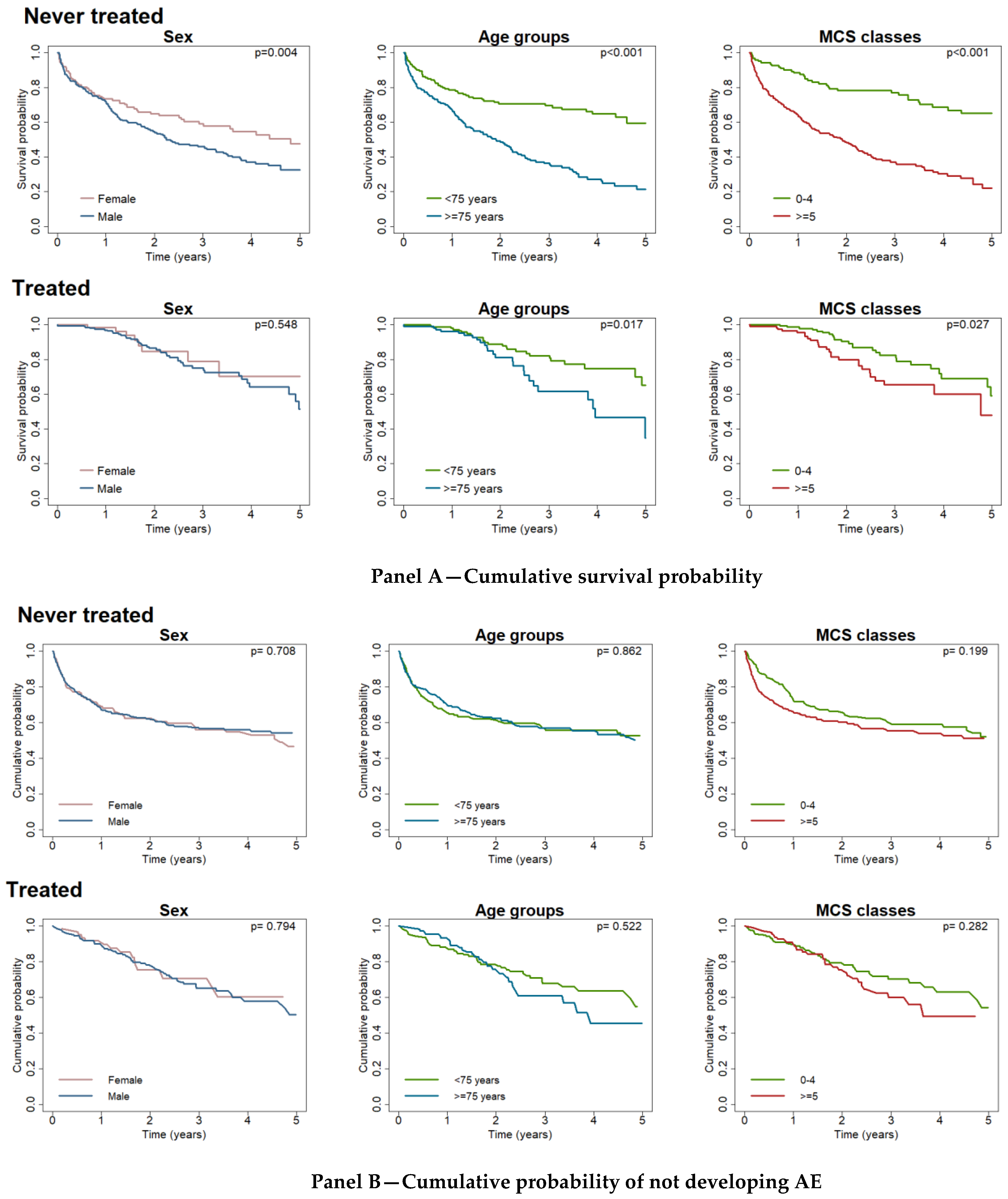
3.2. Survival and Acute Exacerbation Free Cumulative Probability
3.3. Determinants of Acute Exacerbation Event
3.4. Determinants of Mortality
3.5. New Cases of Pulmonary Malignant Neoplasm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.P.; Jenkins, G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, E.R.F.; Daniels, C.E.; Sauver, J.S.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H.; Schroeder, D.R. Incidence, Prevalence, and Clinical Course of Idiopathic Pulmonary Fibrosis: A Population-Based Study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caminati, A.; Madotto, F.; Conti, S.; Cesana, G.; Mantovani, L.; Harari, S. The natural history of idiopathic pulmonary fibrosis in a large European population: The role of age, sex and comorbidities. Intern. Emerg. Med. 2021, 16, 1793–1802. [Google Scholar] [CrossRef]
- Iommi, M.; Bonifazi, M.; Faragalli, A.; Latini, L.L.; Mei, F.; Spazzafumo, L.; Skrami, E.; Ferrante, L.; Carle, F.; Gesuita, R. Occurrence of Idiopathic Pulmonary Fibrosis in Italy: Latest Evidence from Real-World Data. Int. J. Environ. Res. Public Health 2022, 19, 2510. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Kim, H.J.; Perlman, D.; Tomic, R. Natural history of idiopathic pulmonary fibrosis. Respir. Med. 2015, 109, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Collard, H.R.; Yow, E.; Richeldi, L.; Anstrom, K.J.; Glazer, C. For the IPFnet investigators Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir. Res. 2013, 14, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.W.; Hong, S.-B.; Lim, C.-M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2010, 37, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.-A.W.; Dobelle, M.; Padilla, M.; Agovino, M.; Wisnivesky, J.P.; Hashim, D.; Boffetta, P. Idiopathic Pulmonary Fibrosis and Lung Cancer. A Systematic Review and Meta-analysis. Ann. Am. Thorac. Soc. 2019, 16, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Cuello-Garcia, C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Strek, M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef] [Green Version]
- Mooney, J.; Reddy, S.R.; Chang, E.; Broder, M.S.; Gokhale, S.; Corral, M. Antifibrotic therapies reduce mortality and hospitalization among Medicare beneficiaries with idiopathic pulmonary fibrosis. J. Manag. Care Spec. Pharm. 2021, 27, 1724–1733. [Google Scholar] [CrossRef]
- Maher, T.M.; Molina-Molina, M.; Russell, A.-M.; Bonella, F.; Jouneau, S.; Ripamonti, E.; Axmann, J.; Vancheri, C. Unmet needs in the treatment of idiopathic pulmonary fibrosis―insights from patient chart review in five European countries. BMC Pulm. Med. 2017, 17, 124. [Google Scholar] [CrossRef] [Green Version]
- Petnak, T.; Lertjitbanjong, P.; Thongprayoon, C.; Moua, T. Impact of Antifibrotic Therapy on Mortality and Acute Exacerbation in Idiopathic Pulmonary Fibrosis. Chest 2021, 160, 1751–1763. [Google Scholar] [CrossRef]
- Skrami, E.; Carle, F.; Villani, S.; Borrelli, P.; Zambon, A.; Corrao, G.; Trerotoli, P.; Guardabasso, V.; Gesuita, R. Availability of Real-World Data in Italy: A Tool to Navigate Regional Healthcare Utilization Databases. Int. J. Environ. Res. Public Health 2019, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Corrao, G.; Rea, F.; Di Martino, M.; De Palma, R.; Scondotto, S.; Fusco, D.; Lallo, A.; Belotti, L.M.B.; Ferrante, M.; Addario, S.P.; et al. Developing and validating a novel multisource comorbidity score from administrative data: A large population-based cohort study from Italy. BMJ Open 2017, 7, e019503. [Google Scholar] [CrossRef]
- Dong, H.; Robison, L.L.; Leisenring, W.M.; Martin, L.J.; Armstrong, G.T.; Yasui, Y. Estimating the Burden of Recurrent Events in the Presence of Competing Risks: The Method of Mean Cumulative Count. Am. J. Epidemiol. 2015, 181, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Lin, D.Y. Nonparametric analysis of recurrent events and death. Biometrics 2000, 56, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Spagnolo, P.; Bonniaud, P.; Nolin, M.; Dalon, F.; Kirchgässler, K.-U.; Kamath, T.V.; Van Ganse, E.; Belhassen, M. Mortality and Respiratory-Related Hospitalizations in Idiopathic Pulmonary Fibrosis Not Treated With Antifibrotics. Front. Med. 2021, 8, 2815. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, T.M.; Sangaralingham, L.R.; Yao, X.; Sanghavi, D.; Shah, N.D.; Limper, A.H. Clinical Effectiveness of Antifibrotic Medications for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 168–174. [Google Scholar] [CrossRef]
- Sugino, K.; Nakamura, Y.; Ito, T.; Isshiki, T.; Sakamoto, S.; Homma, S. Comparison of clinical characteristics and outcomes between combined pulmonary fibrosis and emphysema associated with usual interstitial pneumonia pattern and non-usual interstitial pneumonia. Sarcoidosis Vasc. Diffus. lung Dis. Off. J. WASOG 2015, 32, 129–137. [Google Scholar]
- Schupp, J.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [Green Version]
- Ohshimo, S.; Ishikawa, N.; Horimasu, Y.; Hattori, N.; Hirohashi, N.; Tanigawa, K.; Kohno, N.; Bonella, F.; Guzman, J.; Costabel, U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Mura, M.; Porretta, M.A.; Bargagli, E.; Sergiacomi, G.; Zompatori, M.; Sverzellati, N.; Taglieri, A.; Mezzasalma, F.; Rottoli, P.; Saltini, C.; et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: A 3-year prospective study. Eur. Respir. J. 2012, 40, 101–109. [Google Scholar] [CrossRef]
- Kim, D.S.; Park, J.H.; Park, B.K.; Lee, J.S.; Nicholson, A.G.; Colby, T. Acute exacerbation of idiopathic pulmonary fibrosis: Frequency and clinical features. Eur. Respir. J. 2006, 27, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Johannson, K.A.; Vittinghoff, E.; Lee, K.; Balmes, J.R.; Ji, W.; Kaplan, G.; Kim, D.S.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur. Respir. J. 2013, 43, 1124–1131. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Total | Never-Treated | Treated | p | |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | ||
| Incident cohort | 676 (100%) | 405 (100%) | 271 (100%) | |
| Sex | <0.001 # | |||
| Female | 226 (33.4%) | 163 (40.2%) | 63 (23.2%) | |
| Male | 450 (66.6%) | 242 (59.8%) | 208 (76.8%) | |
| Median age (IQR) in years | 75 (68–80) | 77 (67–83) | 74 (69–77) | <0.001 * |
| Age group | <0.001 # | |||
| <75 years | 325 (48.1%) | 166 (41%) | 159 (58.7%) | |
| ≥75 years | 351 (51.9%) | 239 (59%) | 112 (41.3%) | |
| MCS classes | <0.001 # | |||
| MCS 0–4 | 286 (42.3%) | 129 (31.9%) | 157 (57.9%) | |
| MCS ≥5 | 390 (57.7%) | 276 (68.1%) | 114 (42.1%) | |
| Mean Cumulative Counts of AEs per 100 patients at | ||||
| 1 year | 48.3 | 56.7 | 33.1 | 0.028 ° |
| 2 years | 60.1 | 67.0 | 49.3 | 0.013 ° |
| 3 years | 68.7 | 73.3 | 61.3 | 0.027 ° |
| 4 years | 72.5 | 75.6 | 68.9 | 0.048 ° |
| 5 years | 78.7 | 80.6 | 75.5 | 0.080 ° |
| Never-Treated | Treated | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Sex (Female r.c.) | ||||||
| Male | 0.98 | 0.73–1.34 | 0.922 | 1.12 | 0.61–2.07 | 0.711 |
| Age groups (<75 r.c.) | ||||||
| ≥75 years | 1.25 | 0.91–1.71 | 0.168 | 1.14 | 0.70–1.87 | 0.593 |
| MCS classes (0–4 r.c.) | ||||||
| ≥5 | 1.61 | 1.14–2.26 | 0.006 | 1.32 | 0.82–2.12 | 0.261 |
| Never-Treated | Treated | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Sex (Female r.c.) | ||||||
| Male | 1.43 | 1.08–1.90 | 0.012 | 1.68 | 0.76–3.71 | 0.202 |
| Age groups (<75 r.c.) | ||||||
| ≥75 years | 2.14 | 1.58–2.91 | <0.001 | 1.88 | 1.07–3.31 | 0.029 |
| MCS classes (0–4 r.c.) | ||||||
| ≥5 | 2.24 | 1.58–3.17 | <0.001 | 1.46 | 0.83–2.57 | 0.190 |
| N. of AE (no AE r.c.) | ||||||
| 1 | 7.02 | 4.92–10.03 | <0.001 | 16.95 | 7.80–36.84 | <0.001 |
| 2 | 8.28 | 5.19–13.20 | <0.001 | 16.54 | 6.93–39.48 | <0.001 |
| 3 or more | 7.03 | 3.93–12.58 | <0.001 | 17.90 | 7.34–43.68 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iommi, M.; Faragalli, A.; Bonifazi, M.; Mei, F.; Latini, L.L.; Pompili, M.; Carle, F.; Gesuita, R. Prognosis and Survival in Idiopathic Pulmonary Fibrosis in the Era of Antifibrotic Therapy in Italy: Evidence from a Longitudinal Population Study Based on Healthcare Utilization Databases. Int. J. Environ. Res. Public Health 2022, 19, 16689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph192416689
Iommi M, Faragalli A, Bonifazi M, Mei F, Latini LL, Pompili M, Carle F, Gesuita R. Prognosis and Survival in Idiopathic Pulmonary Fibrosis in the Era of Antifibrotic Therapy in Italy: Evidence from a Longitudinal Population Study Based on Healthcare Utilization Databases. International Journal of Environmental Research and Public Health. 2022; 19(24):16689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph192416689
Chicago/Turabian StyleIommi, Marica, Andrea Faragalli, Martina Bonifazi, Federico Mei, Lara Letizia Latini, Marco Pompili, Flavia Carle, and Rosaria Gesuita. 2022. "Prognosis and Survival in Idiopathic Pulmonary Fibrosis in the Era of Antifibrotic Therapy in Italy: Evidence from a Longitudinal Population Study Based on Healthcare Utilization Databases" International Journal of Environmental Research and Public Health 19, no. 24: 16689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph192416689