Plant-Pathogen Interaction, Circadian Rhythm, and Hormone-Related Gene Expression Provide Indicators of Phytoplasma Infection in Paulownia fortunei


Abstract
:1. Introduction
2. Results
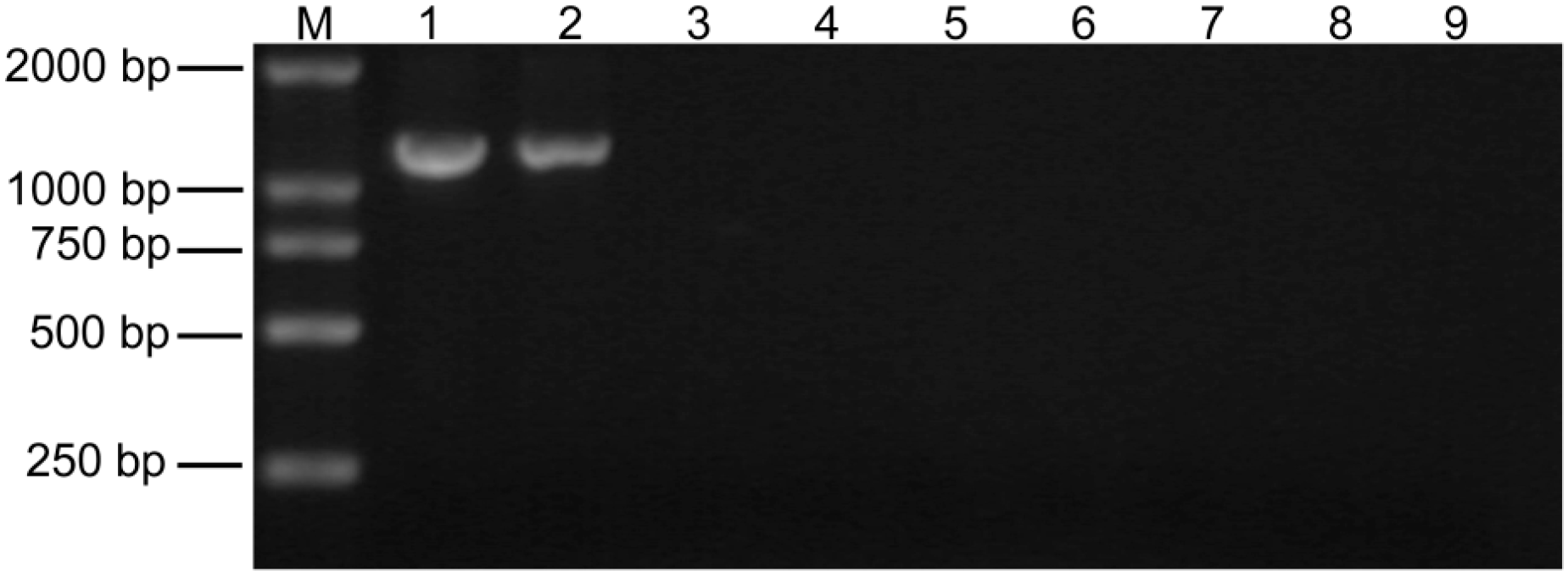
2.1. Detection of Paulownia Witches’ Broom (PaWB) Phytoplasma in Paulownia Showing Disease Symptoms


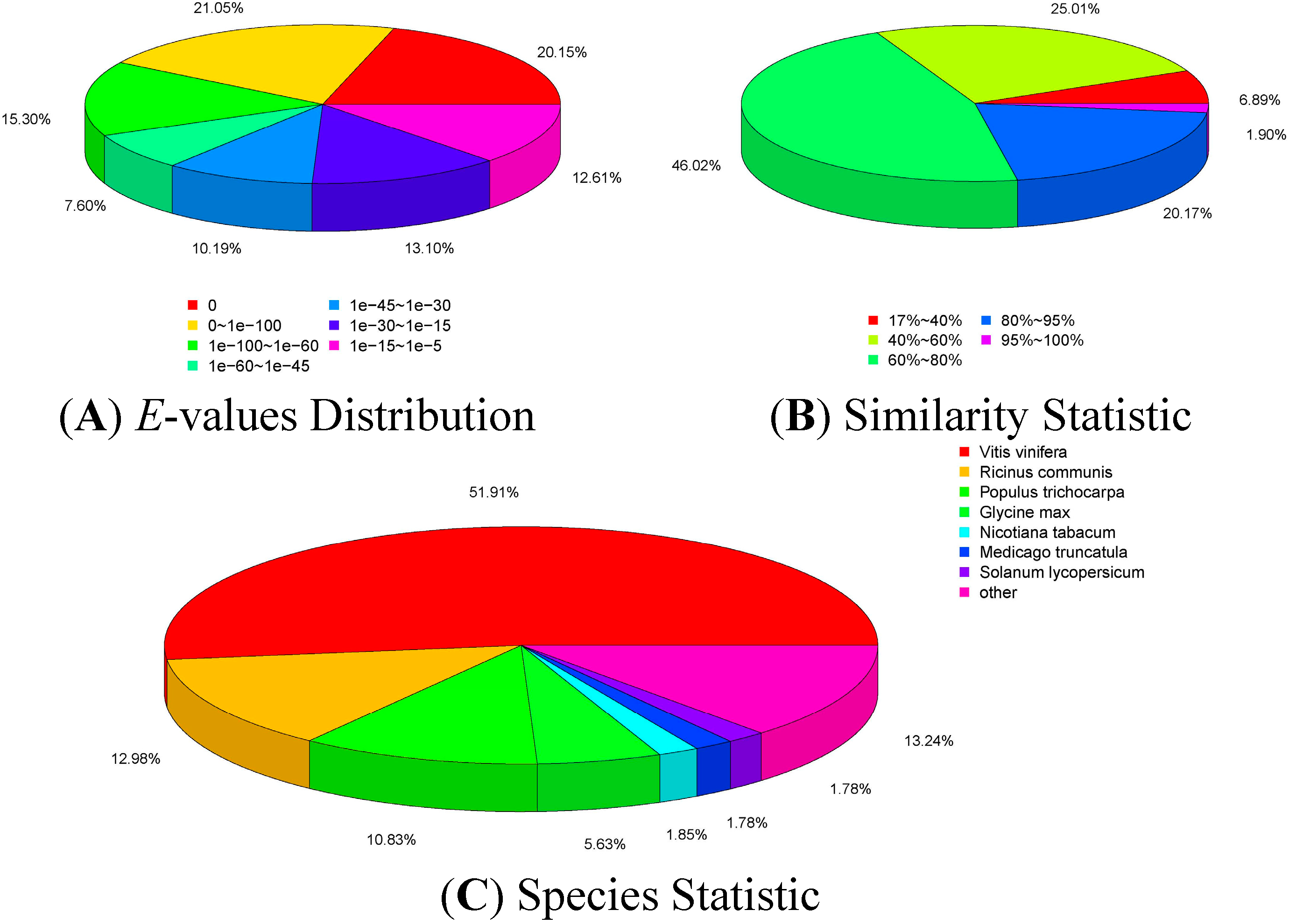
2.2. Transcriptome Sequencing and Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics of Data Production | PF | PFI | PFI-60 |
|---|---|---|---|
| Number of high-quality reads | 57,846,728 | 58,451,828 | 51,417,240 |
| GC percentage (%) | 46.30% | 46.39% | 46.39% |
| Contigs | PF | PFI | PFI-60 |
| Number of contigs | 174,105 | 155,253 | 175,247 |
| Average length of contigs (nt) | 358 | 361 | 339 |
| Length of N50 (bp) | 670 | 740 | 654 |
| Unigenes | PF | PFI | PFI-60 |
| Number of unigenes | 116,749 | 91,997 | 103,300 |
| Length of N50 (bp) | 1963 | 1715 | 1899 |
| Average length of unigenes (bp) | 1016 | 864 | 1029 |
| All unigenes | |||
| Number of all unigenes | 115,116 | ||
| Length of N50 (bp) | 2067 | ||
| Average length of all unigenes (bp) | 1282 |
2.3. Annotation of Non-Redundant Unigenes
| Database | Number of Annotated Unigenes | Percentage of Annotated Unigenes (%) |
|---|---|---|
| nr | 76,507 | 66.5 |
| nt | 69,981 | 60.8 |
| Swiss-Prot | 49,385 | 42.9 |
| KEGG | 46,381 | 40.3 |
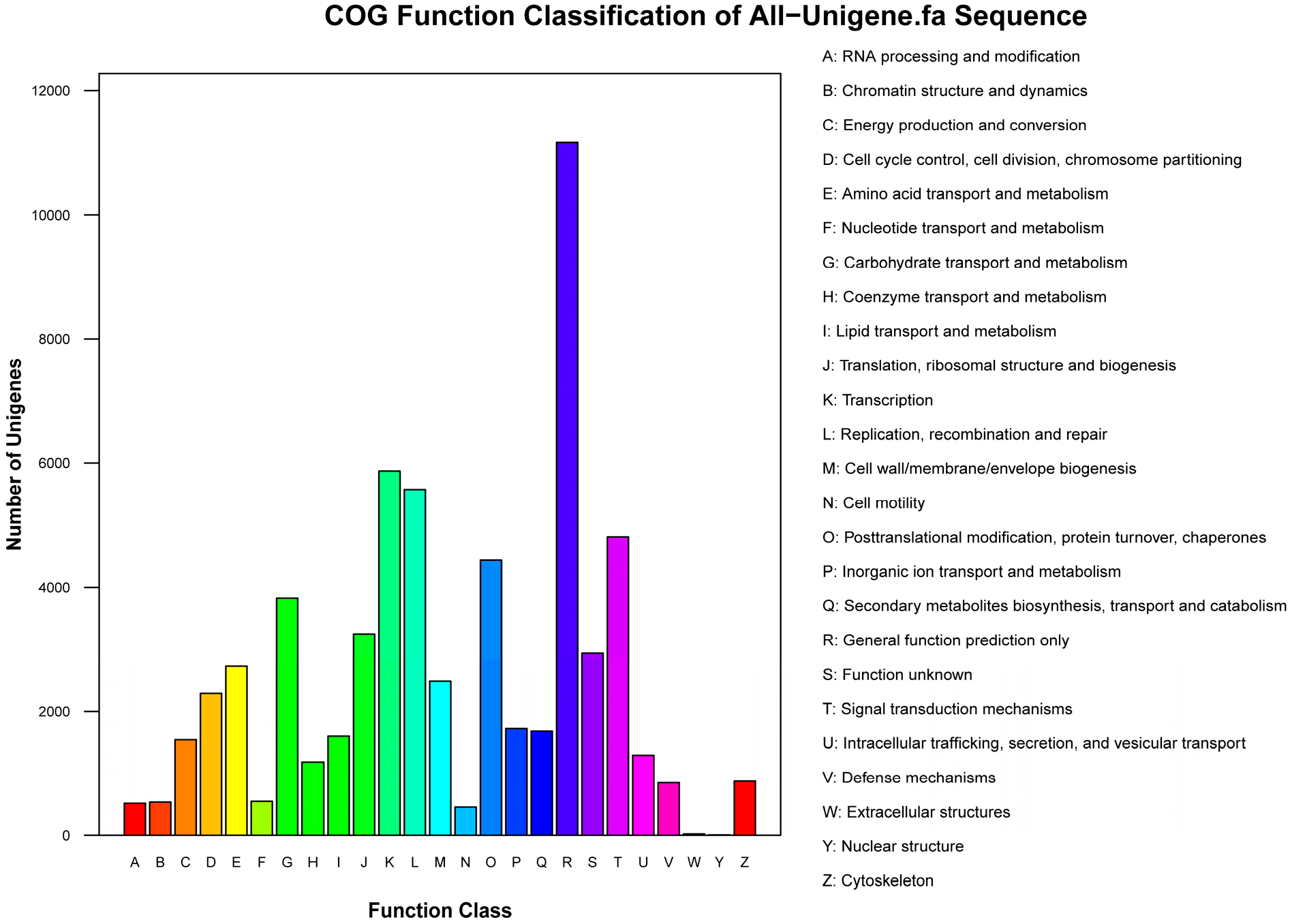
| COG | 32,269 | 28.0 |
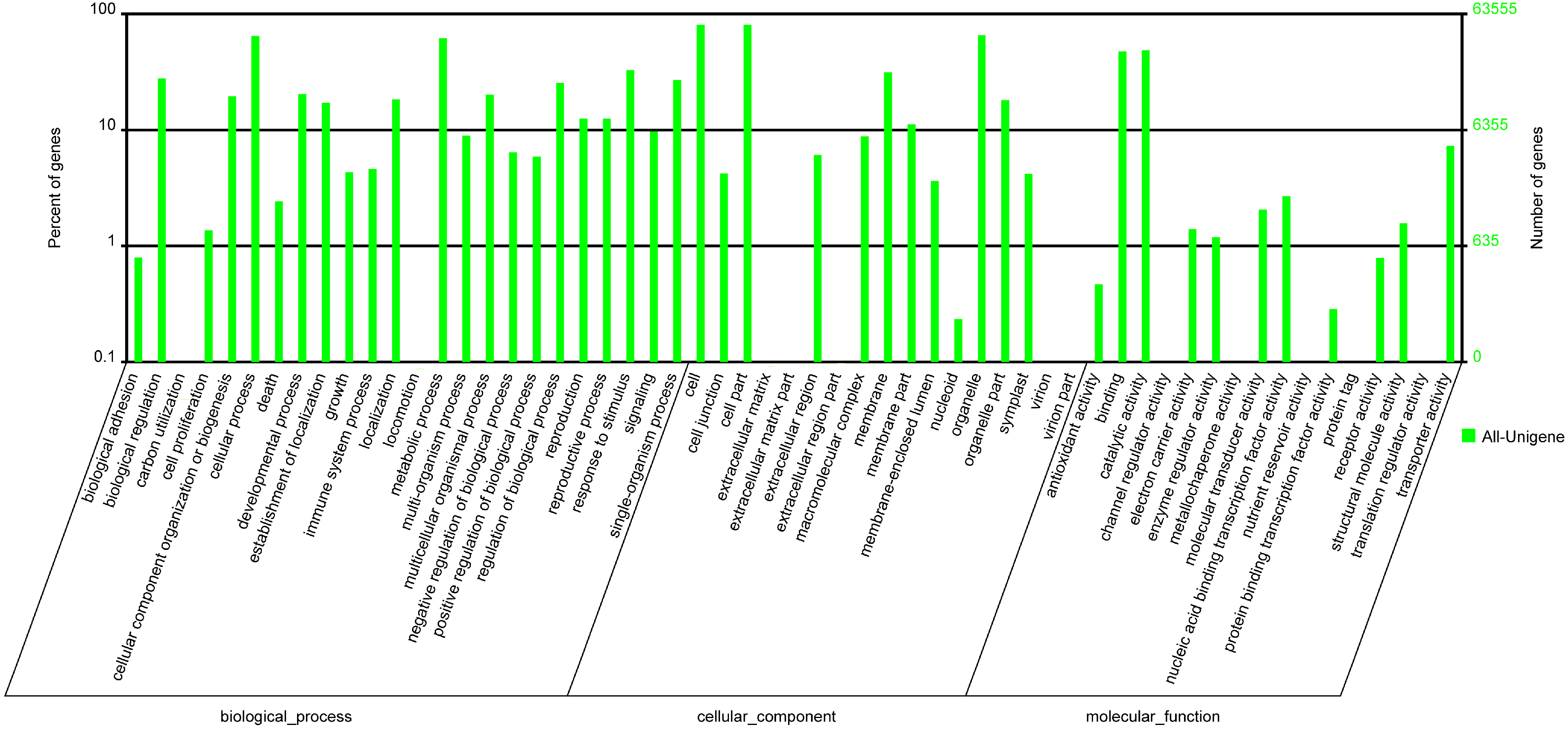
| GO | 63,555 | 55.2 |
| All | 79,035 | 68.7 |



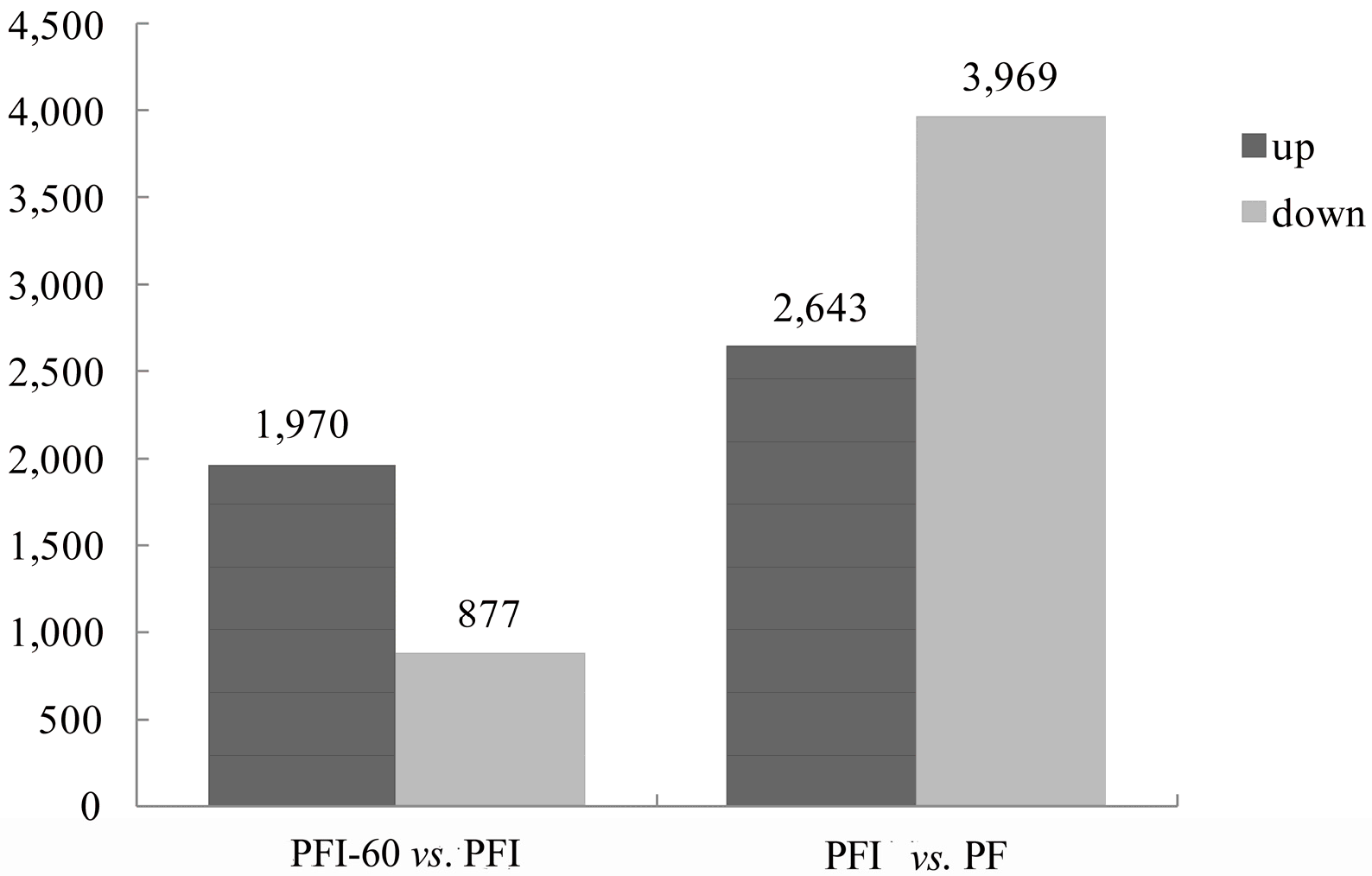
2.4. Functional Classification of the Differentially Expressed Genes (DEGs)

| Gene ID | Gene Length (bp) | Up/Down * | Potential Gene Function |
|---|---|---|---|
| CL10743.Contig1 | 2028 | Up | Inositol-3-phosphate synthase |
| CL10743.Contig2 | 1969 | Up | Inositol-3-phosphate synthase |
| CL10796.Contig1 | 867 | Up | AT5G14920 |
| CL11352.Contig1 | 946 | Up | MLP-like protein 34 |
| CL3131.Contig1 | 2021 | Up | Uncharacterized protein LOC100260696 |
| CL6971.Contig3 | 1890 | Up | Cytochrome P450 |
| CL7523.Contig2 | 721 | Up | Gibberellin induced protein |
| Unigene10760 | 611 | Up | Plasma intrinsic protein 2.2 |
| Unigene12341 | 1172 | Up | Gibberellin induced protein |
| Unigene8595 | 323 | Up | PIP2 protein |
| Unigene8675 | 1282 | Up | UPF0301 protein Cpha266_0885-like isoform 1 |
| CL13170.Contig1 | 858 | Up | Transcription regulator |
| CL9733.Contig2 | 469 | Up | Glycine-rich protein |
| CL3432.Contig3 | 2432 | Up | Microtubule-associated protein TORTIFOLIA1 |
| CL13388.Contig1 | 370 | Up | Aquaporin PIP2 |
| Unigene44849 | 866 | Up | Cell wall-associated hydrolase |
| CL9691.Contig1 | 1215 | Up | Alpha-expansin 2 |
| Unigene43893 | 856 | Up | Hypothetical protein MTR_8g040260 |
| CL6181.Contig1 | 345 | Up | Major latex-like protein |
| CL3545.Contig3 | 2147 | Up | dof zinc finger protein DOF5.2 |
| CL2544.Contig6 | 850 | Down | Putative glycine-rich RNA binding protein |
| CL33.Contig2 | 2834 | Down | WD repeat-containing protein C2A9.03 |
| CL33.Contig3 | 5536 | Down | WD repeat-containing protein C2A9.03 |
| CL33.Contig4 | 5626 | Down | WD-repeat protein |
| CL33.Contig5 | 2815 | Down | WD repeat-containing protein C2A9.03 |
| CL4119.Contig2 | 1113 | Down | Cytochrome P450 72A1 |
| CL4119.Contig5 | 2185 | Down | CYP72A58 |
| CL4739.Contig1 | 727 | Down | Phylloplanin |
| CL4739.Contig2 | 610 | Down | Phylloplanin |
| Unigene10248 | 4052 | Down | WD repeat-containing protein C2A9.03 |
| Unigene10249 | 1966 | Down | WD repeat-containing protein C2A9.03 |
| Unigene22577 | 953 | Down | Unknown |
| Unigene24555 | 3910 | Down | Unnamed protein product |
| Unigene32594 | 743 | Down | Lipid transfer protein |
| Unigene28962 | 1800 | Down | Glucose-6-phosphate/phosphate translocator 2 |
| Unigene30587 | 4360 | Down | GIGANTEA |
| Unigene30582 | 4161 | Down | GIGANTEA |
| Unigene30581 | 4520 | Down | GIGANTEA |
| CL4575.Contig2 | 2531 | Down | 1-Deoxy-d-xylulose-5-phosphate synthase |
| Unigene23241 | 3053 | Down | WD repeat-containing protein C2A9.03 |
| CL3202.Contig3 | 1081 | Up | WRKY protein WRKY29 |
| Unigene12008 | 1848 | Up | Histidine kinase CRE1 |
| CL8962.Contig1 | 915 | Down | Histidine-containing phosphor transfer protein AHP |
| CL12775.Contig1 | 533 | Up | Abscisic acid receptor PYL |
| CL2960.Contig1 | 818 | Up | ABA responsive element binding factor ABF |
| Unigene27866 | 1130 | Up | Adenylate isopentenyl transferase IPT |
| CL4119.Contig2 | 1848 | Down | Cytokinin trans-hydroxylase CYP735A |
| CL4119.Contig5 | 2185 | Down | Cytokinin trans-hydroxylase CYP735A |
| CL2490.Contig6 | 1644 | Down | Serine/threonine-protein kinase PBS1 |
| CL4203.Contig5 | 2362 | Up | Circadian clock associated 1 CCA1 |
| Unigene30581 | 4520 | Down | GIGANTEA |
| Unigene10760 | 611 | Up | Plasma intrinsic protein 2 |
| Unigene14837 | 413 | Up | Plasma membrane H+-ATPase LilHA2 |
| Unigene44439 | 940 | Up | Serine/threonine-protein kinase MEKK1 |
| CL4362.Contig9 | 2426 | Down | Transcription factor MYC2 |
| Unigene19249 | 1046 | Up | Zeatin O-glucosyl transferase CISZOG |
| CL4203.Contig5 | 2326 | Up | Protein CCA1 |
| CL4362.Contig9 | 2426 | Down | Transcription factor PIF3 |
| CL13348.Contig14 | 3472 | Down | Brassinosteroid insensitive 1-associated receptor kinase1 |
| CL4785.Contig7 | 2179 | Down | Serine/threonine protein kinase PBS1 |
| Unigene27866 | 1130 | Up | Adenylate isopentenyl transferase IPT |
| Unigene19249 | 1046 | Up | Cis-zeatin O-glucosyl transferase CISZOG |
| CL2374.Contig8 | 2053 | Down | Lycopene cyclase CruA |
| Unigene28043 | 937 | Up | Timing of CAB expression 1 TOC1 |
| CL12024.Contig1 | 877 | Up | Zinc finger protein CONSTANS |
| Ontology | Class | Numbers of DEGs |
|---|---|---|
| Biological process | biological adhesion | 32 |
| biological regulation | 225 | |
| cell proliferation | 9 | |
| cellular component organization or biogenesis | 103 | |
| cellular process | 482 | |
| death | 44 | |
| developmental process | 173 | |
| establishment of localization | 119 | |
| growth | 31 | |
| immune system process | 43 | |
| localization | 128 | |
| metabolic process | 413 | |
| multi-organism process | 98 | |
| multicellular organismal process | 171 | |
| negative regulation of biological process | 45 | |
| pigmentation | 2 | |
| positive regulation of biological process | 42 | |
| regulation of biological process | 206 | |
| reproduction | 126 | |
| reproductive process | 126 | |
| response to stimulus | 301 | |
| rhythmic process | 17 | |
| signaling | 58 | |
| sulfur utilization | 2 | |
| Cellular component | cell | 547 |
| cell junction | 21 | |
| cell part | 547 | |
| extracellular matrix | 1 | |
| extracellular region | 78 | |
| macromolecular complex | 55 | |
| membrane | 230 | |
| membrane part | 63 | |
| membrane-enclosed lumen | 41 | |
| organelle | 392 | |
| organelle part | 102 | |
| symplast | 21 | |
| Molecular function | antioxidant activity | 3 |
| binding | 341 | |
| catalytic activity | 307 | |
| electron carrier activity | 22 | |
| enzyme regulator activity | 20 | |
| molecular transducer activity | 13 | |
| nucleic acid binding transcription factor activity | 25 | |
| nutrient reservoir activity | 1 | |
| protein binding transcription factor activity | 3 | |
| receptor activity | 3 | |
| structural molecule activity | 2 | |
| transporter activity | 53 |
| # | Pathway | DEGs with Pathway Annotation | p-Value | Q Value | Pathway ID |
|---|---|---|---|---|---|
| 1 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 30 (5.61%) | 2.32 × 10−18 | 1.92 × 10−16 | ko00945 |
| 2 | Limonene and pinene degradation | 22 (4.11%) | 2.20 × 10−13 | 9.13 × 10−12 | ko00903 |
| 3 | Circadian rhythm-plant | 30 (5.61%) | 3.34 × 10−12 | 9.23 × 10−11 | ko04712 |
| 4 | RNA degradation | 35 (6.54%) | 3.30 × 10−11 | 6.85 × 10−10 | ko03018 |
| 5 | Carotenoid biosynthesis | 21 (3.93%) | 8.94 × 10−10 | 1.48 × 10−8 | ko00906 |
| 6 | Biosynthesis of secondary metabolites | 92 (17.2%) | 3.82 × 10−7 | 5.29 × 10−6 | ko01110 |
| 7 | Tryptophan metabolism | 11 (2.06%) | 2.70 × 10−6 | 3.21 × 10−5 | ko00380 |
| 8 | Porphyrin and chlorophyll metabolism | 15 (2.8%) | 6.78 × 10−6 | 6.85 × 10−5 | ko00860 |
| 9 | Monoterpenoid biosynthesis | 7 (1.31%) | 7.43 × 10−6 | 6.85 × 10−5 | ko00902 |
| 10 | Isoflavonoid biosynthesis | 6 (1.12%) | 3.32 × 10−5 | 2.76 × 10−4 | ko00943 |
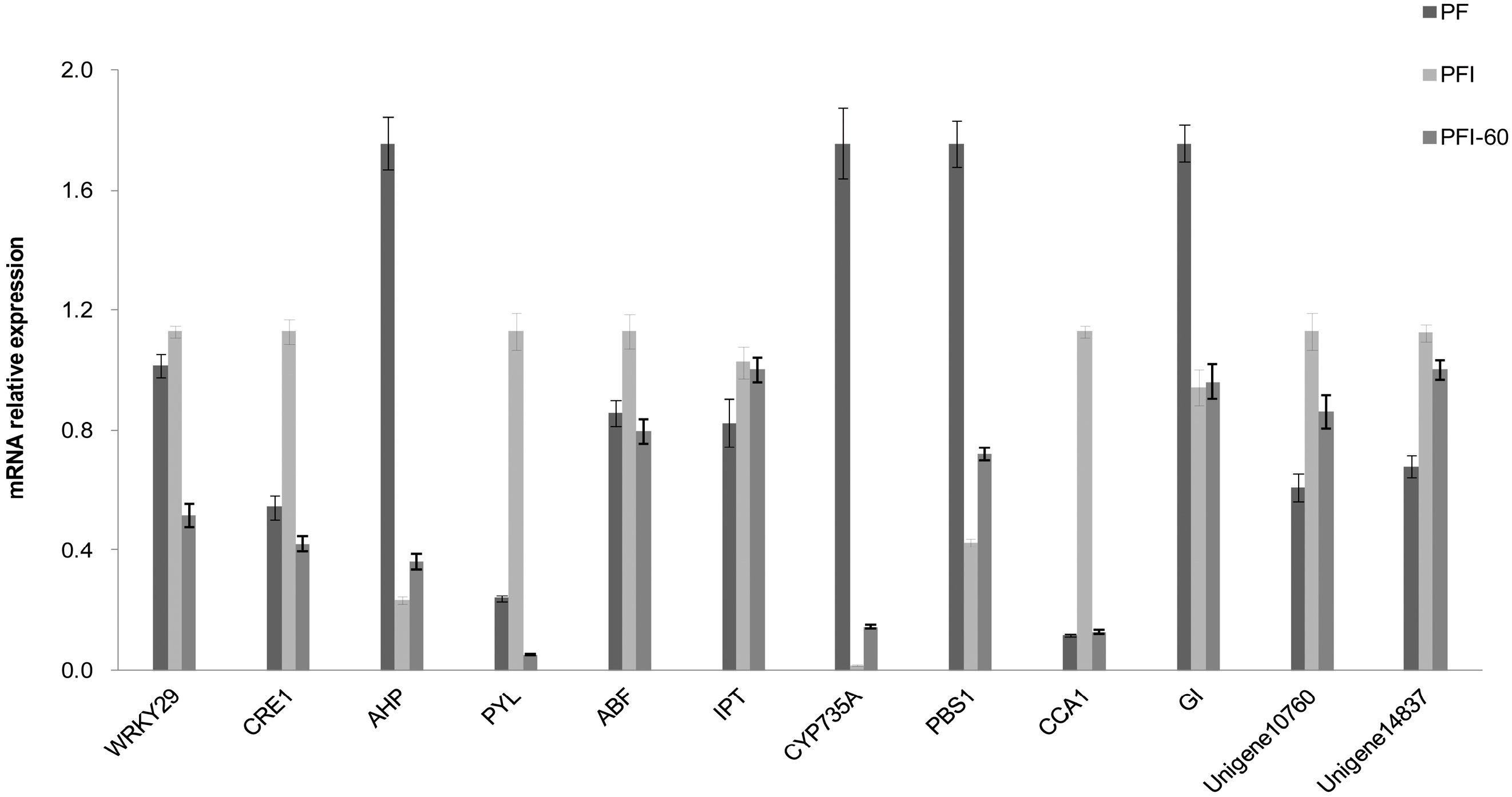
2.5. Confirmation of the RNA-Seq Expression Data by qRT-PCR

3. Discussion
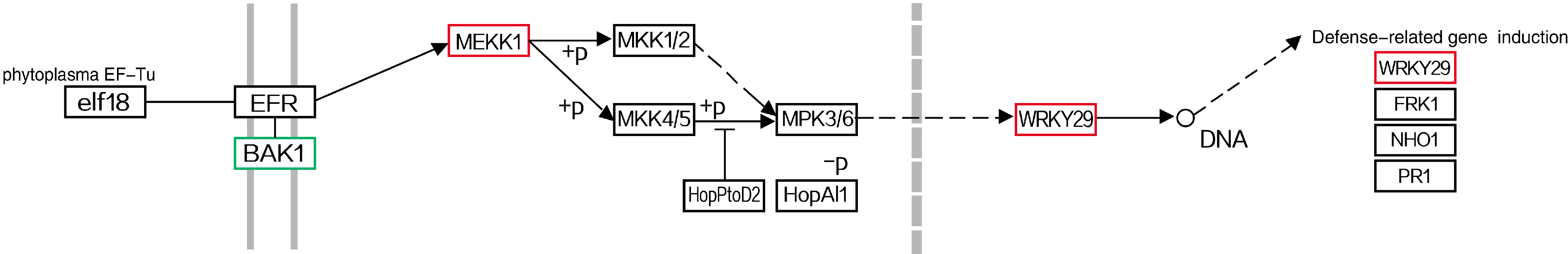
3.1. DEGs Related to Plant-Pathogen Interaction

3.2. DEGs Related to Hormone Biosynthesis
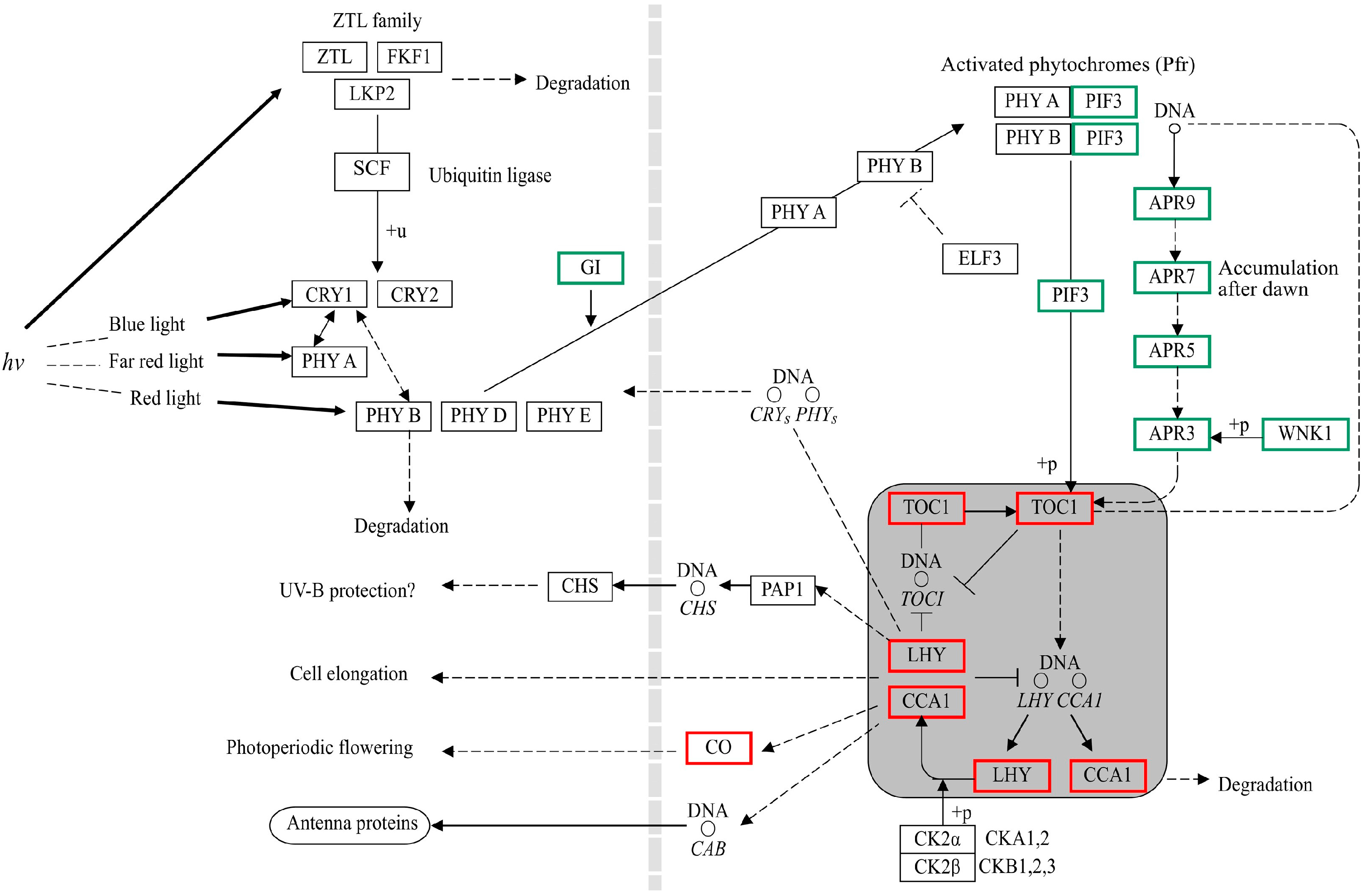
3.3. DEGs Related to Circadian Rhythms

4. Experimental Section
4.1. Preparation of Plant Material
4.2. Construction of cDNA Libraries of P. fortunei
4.3. Bioinformatics Analysis
4.4. Database Searches
4.5. Unigene Gene Ontology (GO) Classification
4.6. Protein Coding Sequence (CDS) Prediction
4.7. Unigene Expression Analysis
4.8. GO Functional Enrichment Analysis of the DEGs
4.9. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis of the DEGs
4.10. Quantitative Real-time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakamura, H.; Yoshikawa, N.; Takahashi, T.; Sahashi, N.; Kubono, T.; Shoji, T. Evaluation of primer pairs for the reliable diagnosis of Paulownia witches’-broom disease using a polymerase chain reaction. Plant Dis. 1996, 80, 302–305. [Google Scholar] [CrossRef]
- Hiruki, C. Paulownia witches’-broom disease important in East Asia. Int. Symp. Urban Tree Health 1997, 469, 63–68. [Google Scholar]
- Namba, S. Molecular biological studies on phytoplasmas. J. Gen. Plant Pathol. 2002, 68, 257–259. [Google Scholar]
- Bayliss, K.L.; Saqib, M.; Dell, B.; Jones, M.G.K.; Hardy, G.E.S.J. First record of “Candidatus Phytoplasma australiense” in Paulownia trees. Australas. Plant Path. 2005, 34, 123–124. [Google Scholar] [CrossRef]
- Doi, M.; Tetranaka, M.; Yora, K.; Asuyama, H. Mycoplasma or PLTgroup-like organisms found in the phloem elements of plants infected with mulberry dwarf, potato witches’ broom, aster yellows or Paulownia witches’ broom. Ann. Phytopathol. Soc. Jpn. 1967, 33, 259–266. [Google Scholar] [CrossRef]
- Group., T.I.P.; Firrao, G. “Candidatus Phytoplasma”,a taxon for the wall-less, nonhelical prokaryotes that colonize plant phloem and insects. Int. J. Syst. Evol. Microbiol. 2004, 54, 1243–1255. [Google Scholar] [PubMed]
- Fan, G.; Zhang, S.; Zhai, X.; Liu, F.; Dong, Z. Effects of antibiotics on the Paulownia witches’ broom phytoplasmas and pathogenic protein related to witches’ broom symptom. Sci. Silv. Sin. 2007, 43, 138–142. [Google Scholar]
- Zhai, X.; Cao, X.; Fan, G. Growth of Paulownia witche’ broom seedlings treated with methyl methane sulphonate and SSR analysis. Sci. Silv. Sin. 2010, 46, 176–181. [Google Scholar]
- Cao, X.; Fan, G.; Deng, M.; Zhao, Z.; Dong, Y. Identification of genes related to Paulownia witche’ broom by AFLP and MSAP. Int. J. Mol. Sci. 2014, 15, 14669–14683. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Botti, S.; Caprara, L.; Arthofer, W.; Strommer, S.; Hanzer, V.; Katinger, H.; Bertaccini, A.; Machado, M.L.D.C. Improved detection methods for fruit tree phytoplasmas. Plant Mol. Biol. Rep. 2001, 19, 169–179. [Google Scholar] [CrossRef]
- Baric, S.; Kerschbamer, C.; Dalla Via, J. TaqMan real-time PCR vs. four conventional PCR assays for detection of apple proliferation phytoplasma. Plant Mol. Biol. Rep. 2006, 24, 169–184. [Google Scholar] [CrossRef]
- Mou, H.Q.; Lu, J.; Zhu, S.F.; Lin, C.L.; Tian, G.Z.; Xu, X.; Zhao, W.J. Transcriptomic analysis of Paulownia infected by Paulownia witches’-broom Phytoplasma. PLoS One 2013, 8, e77217. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Dong, Y.; Fan, G.; Zhao, Z.; Deng, M.; Cao, X.; Niu, S. Discovery of genes related to witches broom disease in Paulownia tomentosa × Paulownia fortunei by a de novo assembled transcriptome. PLoS One 2013, 8, e80238. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Hammond, R.; Davis, R.; Gundersen, D. Universal amplification and analysis of pathogen 16S rDNA for classification and identification of mycoplasmalike organisms. Phytopathology 1993, 83, 834–842. [Google Scholar] [CrossRef]
- Durrant, W.E.; Rowland, O.; Piedras, P.; Hammond-Kosack, K.E.; Jones, J.D. cDNA–AFLP reveals a striking overlap in race-specific resistance and wound response gene expression profiles. Plant Cell Online 2000, 12, 963–977. [Google Scholar] [CrossRef]
- Xiong, L.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14, S165–S183. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Kakizawa, S.; Nishigawa, H.; Jung, H.Y.; Wei, W.; Suzuki, S.; Arashida, R.; Nakata, D.; Miyata, S.I.; Ugaki, M. Reductive evolution suggested from the complete genome sequence of a plant-pathogenic phytoplasma. Nat. Genet. 2004, 36, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Schwessinger, B.; Zipfel, C. News from the frontline: Recent insights into PAMP-triggered immunity in plants. Curr. Opin. Plant Biol. 2008, 11, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C.; Kunze, G.; Chinchilla, D.; Caniard, A.; Jones, J.D.; Boller, T.; Felix, G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 2006, 125, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Kunze, G.; Zipfel, C.; Robatzek, S.; Niehaus, K.; Boller, T.; Felix, G. The N terminus of bacterial elongation factor Tu elicits innate immunity in Arabidopsis plants. Plant Cell Online 2004, 16, 3496–3507. [Google Scholar] [CrossRef]
- Christensen, N.M.; Axelsen, K.B.; Nicolaisen, M.; Schulz, A. Phytoplasmas and their interactions with hosts. Trends Plant Sci. 2005, 10, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Oshima, K.; Kakizawa, S.; Ishii, Y.; Ozeki, J.; Hashimoto, M.; Komatsu, K.; Kagiwada, S.; Yamaji, Y.; Namba, S. A unique virulence factor for proliferation and dwarfism in plants identified from a phytopathogenic bacterium. Proc. Natl. Acad. Sci. USA 2009, 106, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, K.; Tarkowski, P.; Matsumoto-Kitano, M.; Kato, T.; Sato, S.; Tarkowska, D.; Tabata, S.; Sandberg, G.; Kakimoto, T. Roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16598–16603. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, T. Biosynthesis of cytokinins. J. Plant Res. 2003, 116, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kamada-Nobusada, T.; Sakakibara, H. Molecular basis for cytokinin biosynthesis. Phytochemistry 2009, 70, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Ćurković Perica, M. Auxin-treatment induces recovery of phytoplasma-infected periwinkle. J. Appl. Microbiol. 2008, 105, 1826–1834. [Google Scholar]
- Weintraub, P.G.; Jones, P. Phytoplasmas: Genomes, Plant Hosts and Vectors; CABI: Wallingford, UK, 2010. [Google Scholar]
- Medford, J.I.; Horgan, R.; El-Sawi, Z.; Klee, H.J. Alterations of endogenous cytokinins in transgenic plants using a chimeric isopentenyl transferase gene. Plant Cell Online 1989, 1, 403–413. [Google Scholar] [CrossRef]
- Cline, M.; Wesse, T.; Iwamura, H. Cytokinin/auxin control of apical dominance in Ipomoea nil. Plant Cell Physiol. 1997, 38, 659–667. [Google Scholar] [CrossRef]
- Arney, S.; Mitchell, D. The effect of abscisic acid on stem elongation and correlative inhibition. New Phytol. 1969, 68, 1001–1015. [Google Scholar] [CrossRef]
- Kaufman, P.B.; Jones, R.A. Regulation of growth in Avena (Oat) stem segments by gibberellic acid and abscisic acid. Physiol. Plant. 1974, 31, 39–43. [Google Scholar] [CrossRef]
- Nambara, E.; Marion-Poll, A. Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 2005, 56, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Tepperman, J.M.; Quail, P.H. PIF3, a phytochrome-interacting factor necessary for normal photoinduced signal transduction, is a novel basic helix-loop-helix protein. Cell 1998, 95, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, P.G.; Fankhauser, C.; Terry, M.J. PIF3 is a repressor of chloroplast development. Proc. Natl. Acad. Sci. USA 2009, 106, 7654–7659. [Google Scholar] [CrossRef] [PubMed]
- Soy, J.; Leivar, P.; González-Schain, N.; Sentandreu, M.; Prat, S.; Quail, P.H.; Monte, E. Phytochrome-imposed oscillations in PIF3 protein abundance regulate hypocotyl growth under diurnal light/dark conditions in Arabidopsis. Plant J. 2012, 71, 390–401. [Google Scholar] [PubMed]
- Levy, Y.Y.; Dean, C. Control of flowering time. Curr. Opin. Plant Biol. 1998, 1, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Alabadı́, D.; Oyama, T.; Yanovsky, M.J.; Harmon, F.G.; Mas, P.; Kay, S.A. Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 2001, 293, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tobin, E.M. Constitutive expression of the CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) gene disrupts circadian rhythms and suppresses its own expression. Cell 1998, 93, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, G.; Dong, Y.; Deng, M.; Zhao, Z.; Niu, S.; Xu, E. Plant-Pathogen Interaction, Circadian Rhythm, and Hormone-Related Gene Expression Provide Indicators of Phytoplasma Infection in Paulownia fortunei. Int. J. Mol. Sci. 2014, 15, 23141-23162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223141
Fan G, Dong Y, Deng M, Zhao Z, Niu S, Xu E. Plant-Pathogen Interaction, Circadian Rhythm, and Hormone-Related Gene Expression Provide Indicators of Phytoplasma Infection in Paulownia fortunei. International Journal of Molecular Sciences. 2014; 15(12):23141-23162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223141
Chicago/Turabian StyleFan, Guoqiang, Yanpeng Dong, Minjie Deng, Zhenli Zhao, Suyan Niu, and Enkai Xu. 2014. "Plant-Pathogen Interaction, Circadian Rhythm, and Hormone-Related Gene Expression Provide Indicators of Phytoplasma Infection in Paulownia fortunei" International Journal of Molecular Sciences 15, no. 12: 23141-23162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223141