
Lentiviral-Mediated Short Hairpin RNA Knockdown of MTDHInhibits Cell Growth and Induces Apoptosis by Regulatingthe PTEN/AKT Pathway in Hepatocellular Carcinoma

Abstract
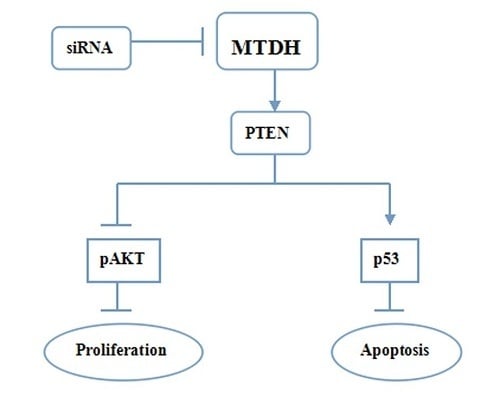
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
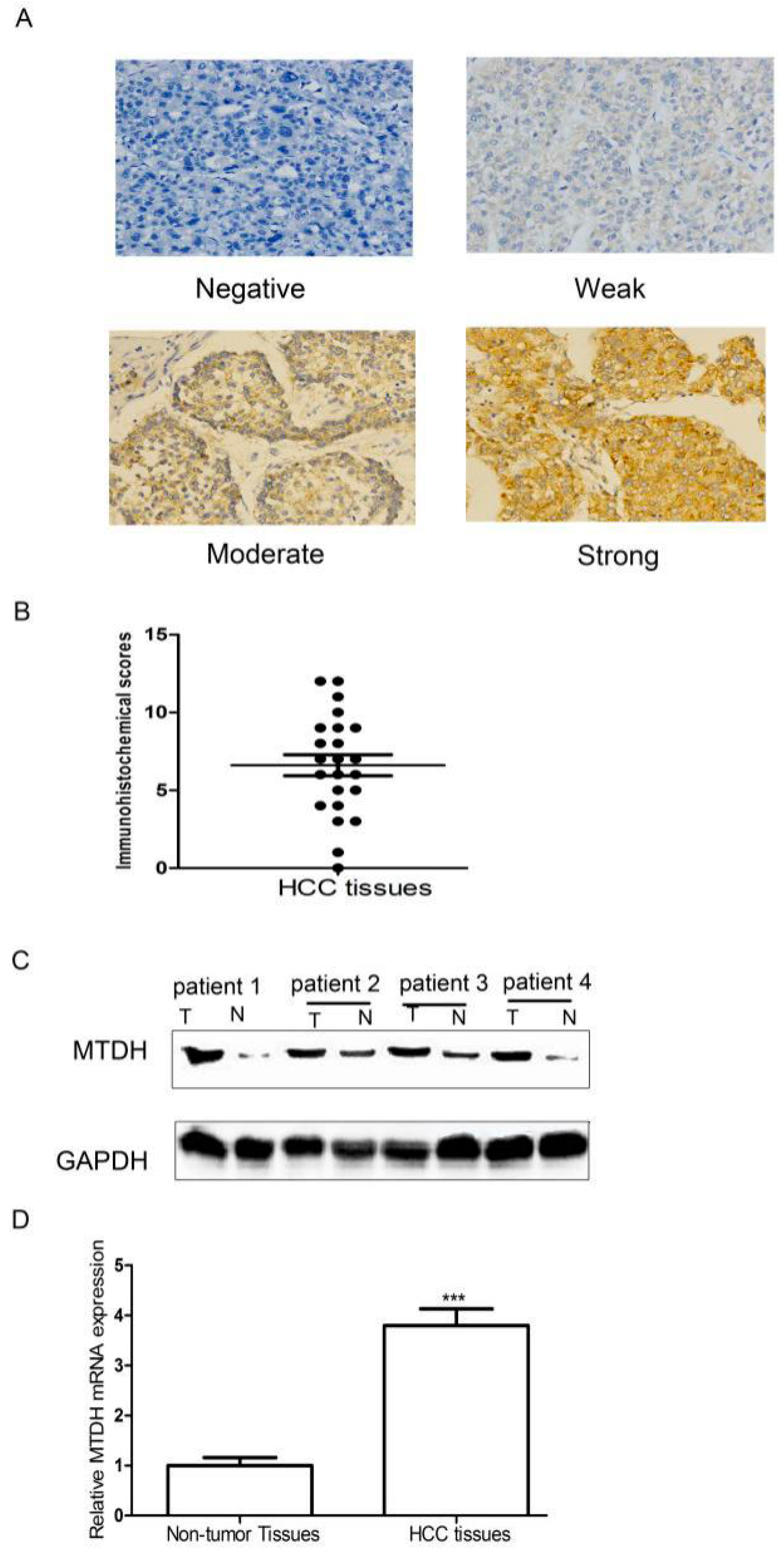
2.1. Expression of MTDH Was Significantly Upregulated in HCC Tissues

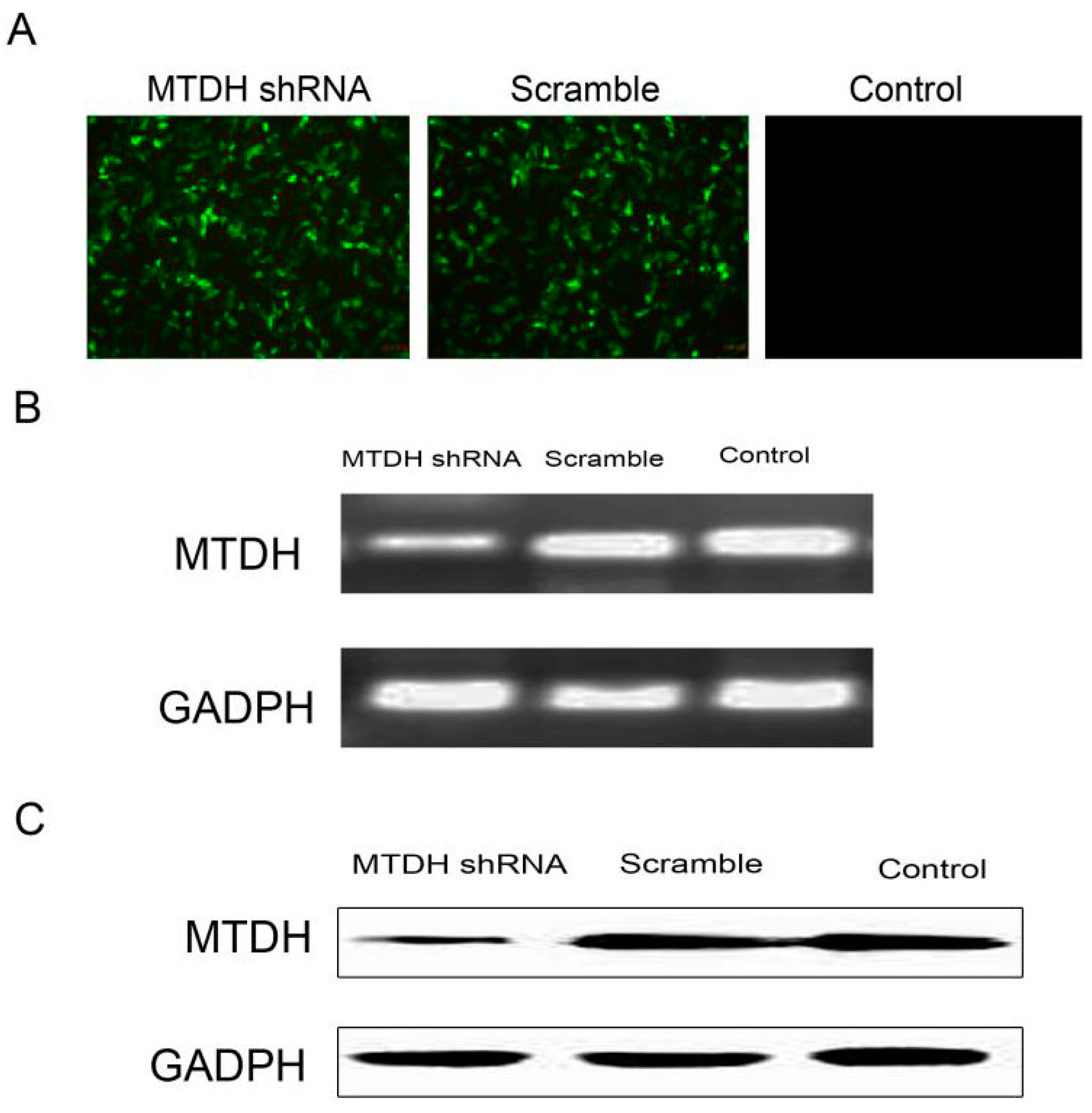
2.2. shRNA Silencing of MTDH Expression Effectively Reduces MTDH mRNA and Protein Expression

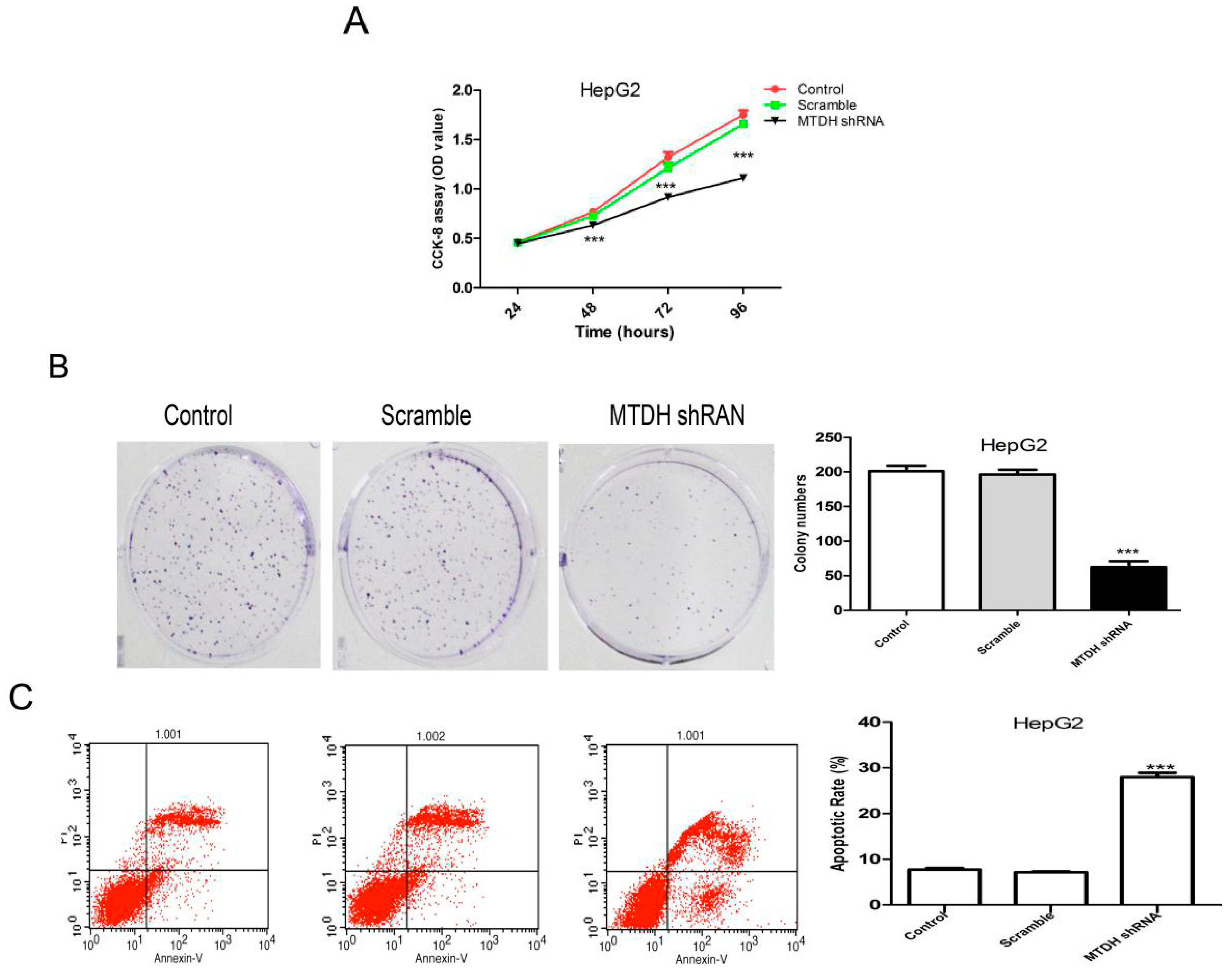
2.3. Knockdown of MTDH Inhibits the Viability, Colony Formation and Induced Apoptosis in HepG2 Cells

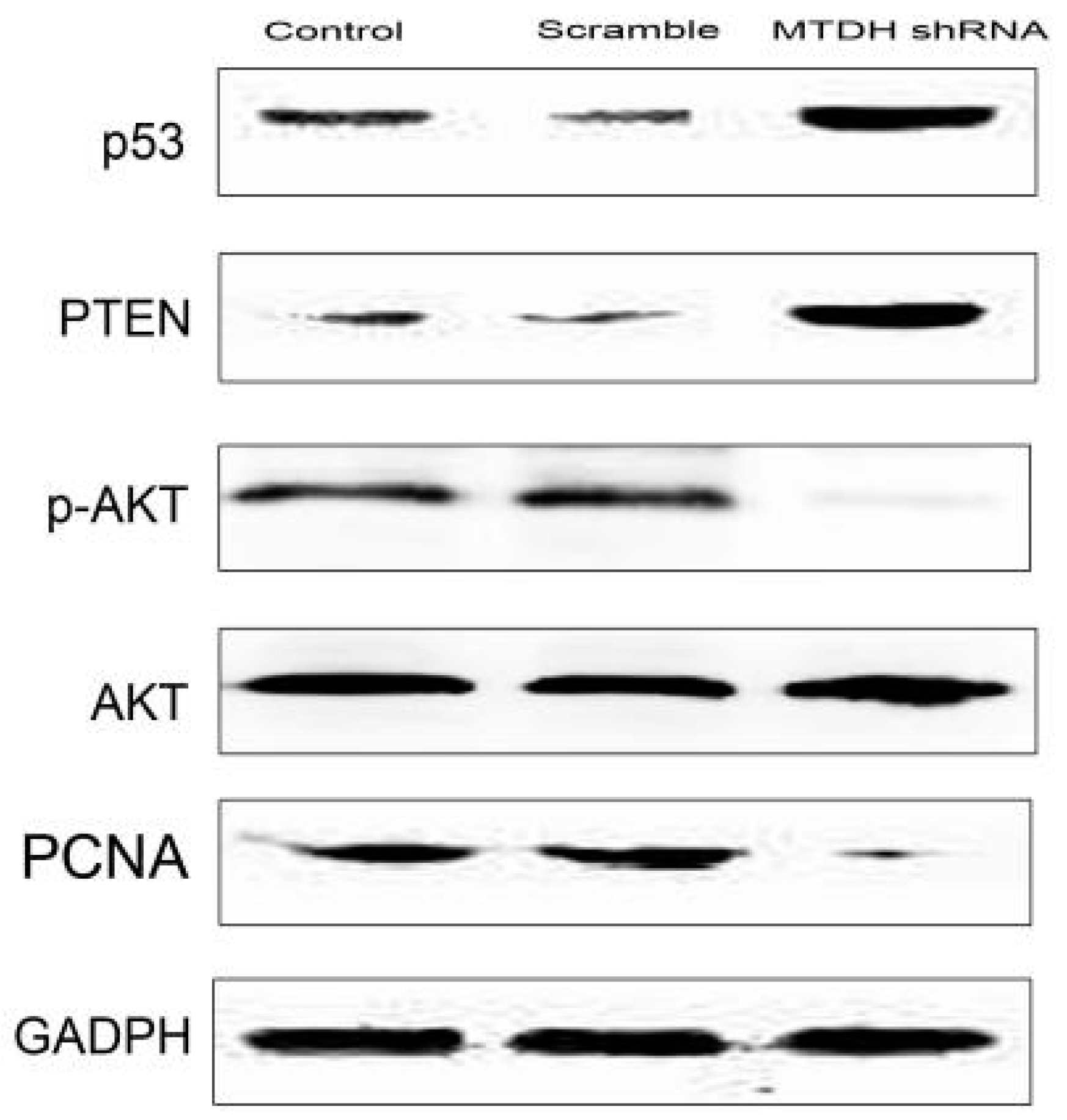
2.4. Knockdown of MTDH Inhibits Phosphorylation of AKT and Increased Apoptosis Related Protein Expression

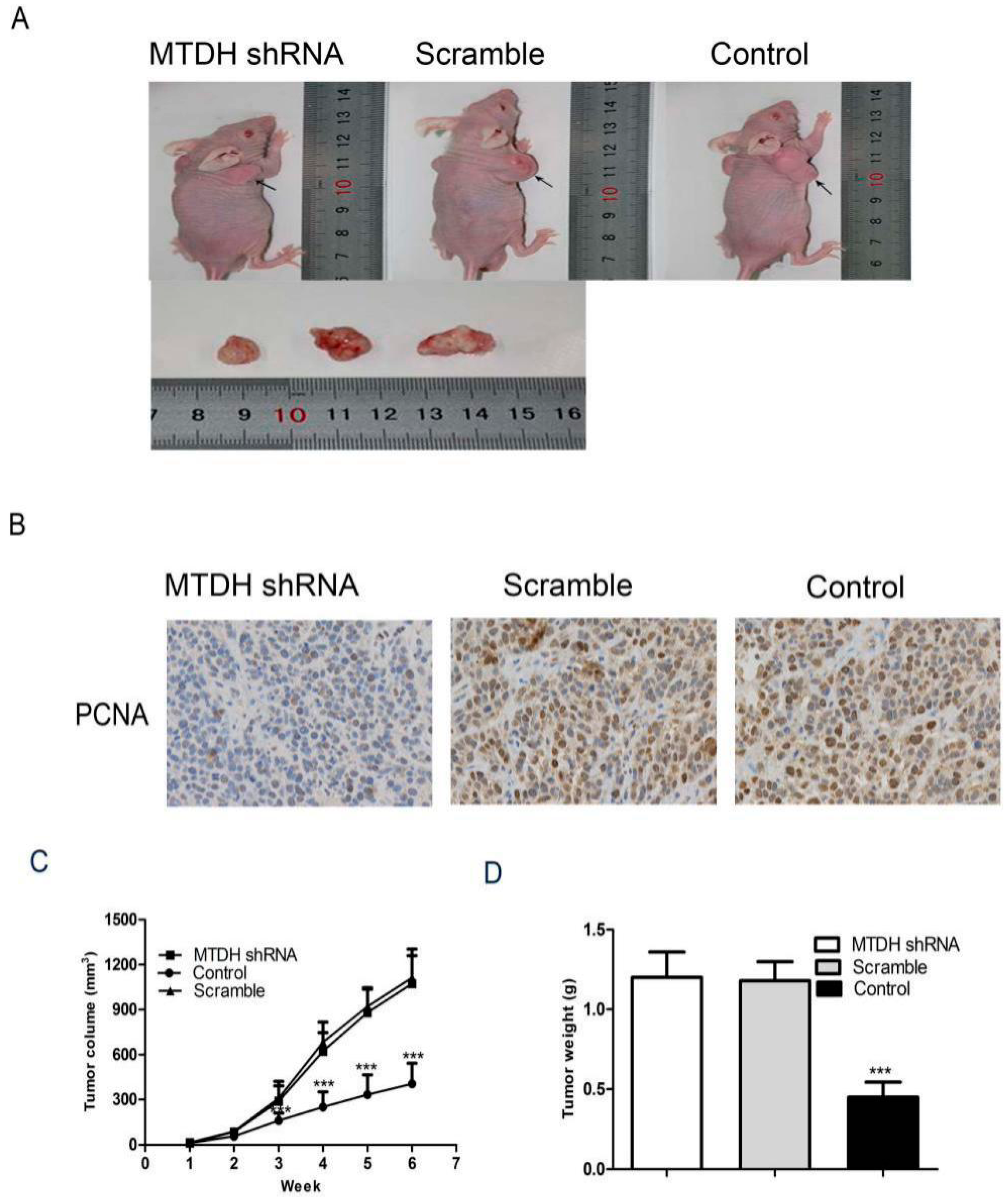
2.5. Knockdown of MTDH Inhibits HepG2 Tumor Growth in Xenograft Model

3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. RT-PCR of MTDH
4.3. Tissue Samples
4.4. Western Blot Analysis
4.5. siRNA-Mediated Silencing of MTDH
4.6. CCK-8 Assay
4.7. Colony Formation Assay
4.8. Cell Apoptosis Assay
4.9. Immunohistochemical Analysis
4.10. Immunohistochemical Results Evaluation
4.11. In Vivo Tumorigenicity
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Freeman, R.B.; Edwards, E.B.; Harper, A.M. Waiting list removal rates among patients with chronic and malignant liver diseases. Am. J. Transplant. 2006, 6, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, D.; Zhang, Y.; Lou, G.; Huang, G.; Chen, B.; Shen, X.; Gao, M.; Gong, W.; Zhou, P.; et al. LXRα-mediated downregulation of FOXM1 suppresses the proliferation of hepatocellular carcinoma cells. Oncogene 2014, 33, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; We, Y.; Kang, Y. The multifaceted role of MTDH/AEG-1 in cancer progression. Clin. Cancer Res. 2009, 15, 5615–5620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, Y.B.; Gao, Y.L.; Yan, B.; Yang, J.R.; Guo, Q. Metadherin in prostate, bladder, and kidney cancer: A systematic review. Mol. Clin. Oncol. 2014, 2, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Liu, W.; You, N.; Wang, T.; Wang, X.; Lu, P.; Zhao, G.; Yang, P.; Wang, D.; Dou, K. Prognostic significance of metadherin overexpression in hepatitis B virus-related hepatocellular carcinoma. Oncol. Rep. 2012, 27, 2073–2079. [Google Scholar] [PubMed]
- Zhu, K.; Dai, Z.; Pan, Q.; Wang, Z.; Yang, G.H.; Yu, L.; Ding, Z.B.; Shi, G.M.; Ke, A.W.; Yang, X.R. Metadherin promotes hepatocellular carcinoma metastasis through induction of epithelial mesenchymal transition. Clin. Cancer Res. 2011, 17, 7294–7302. [Google Scholar] [CrossRef] [PubMed]
- Soomin, A.; Jiyeon, H.; Cheol-Keun, P. Metadherin is a prognostic predictor of hepatocellular carcinoma after curative hepatectomy. Gut Liver 2013, 7, 206–212. [Google Scholar]
- Robertson, C.L.; Srivastava, J.; Siddiq, A.; Gredler, R.; Emdad, L.; Rajasekaran, D.; Akiel, M.; Shen, X.N.; Guo, C.; Giashuddin, S. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res. 2014, 74, 6184–6193. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Siddiq, A.; Emdad, L.; Santhekadur, P.K.; Chen, D.; Gredler, R.; Shen, X.N.; Robertson, C.L.; Dumur, C.I.; Hylemon, P.B.; et al. Astrocyte elevated gene-1 promotes hepatocarcinogenesis: Novel insights from a mouse model. Hepatology 2012, 56, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Lee, C.R.; Man, S.; Chen, E.; Kerbel, R.S. Effects of sorafenib dose on acquired reversible resistance and toxicity in hepatocellular carcinoma. Cancer Res. 2015, 75, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A-L.; Kang, Y-K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Emdad, L.; Lee, S.G.; Su, Z.Z.; Jeon, H.Y.; Boukerche, H.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21300–21305. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Song, L.; Xiong, H.; Wang, L.; Yan, X.; Yuan, J.; Wu, J.; Li, M. Astrocyte elevated gene-1 is a proliferation promoter in breast cancer via suppressing transcriptional factor FOXO1. Oncogene 2009, 28, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.F.; Mao, X.; Zeng, C.; He, S.; Li, S.; Wang, L.T. AEG-1 expression characteristics in human non-small cell lung cancer and its relationship with apoptosis. Med. Oncol. 2013, 30. [Google Scholar] [CrossRef] [PubMed]
- Lokody, I.B.; Francis, J.C.; Gardiner, J.R.; Erler, J.T.; Swain, A. PTEN regulates epithelial cytodifferentiation during prostate development. PLoS ONE 2015, 10, e0129470. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Siraj, A.K.; Prabhakaran, S.; Jehan, Z.; Ajarim, D.; Al-Dayel, F.; Tulbah, A.; Al-Kuraya, K.S. Loss of PTEN expression is associated with aggressive behavior and poor prognosis in Middle Eastern triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 151, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Siraj, A.K.; Jehan, Z.; Prabakaran, S.; Al-Sobhi, S.S.; Al-Dawish, M.; Al-Dayel, F.; Al-Kuraya, K.S. PTEN loss is associated with follicular variant of Middle Eastern papillary thyroid carcinoma. Br. J. Cancer 2015, 112, 1938–1943. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Y.A.; Pangas, S.A.; Adams, J.; Zhou, W.; Castrillon, D.H.; Wilhelm, D.; Richards, J.S. FOXO1/3 and PTEN depletion in granulosa cells promotes ovarian granulosa cell tumor development. Mol. Endocrinol. 2015, 29, 1006–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, X.; Liu, S.; Li, J.; Wen, Z.; Li, M. 17betaE2 promotes cell proliferation in endometriosis by decreasing PTEN via NFkappaB-dependent pathway. Mol. Cell. Endocrinol. 2010, 317, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhao, D.; Hatanpaa, K.J.; Mickey, B.E.; Saha, D.; Boothman, D.A.; Story, M.D.; Wong, E.T.; Burma, S.; Georgescu, M.M.; et al. RIP1 activates PI3K-Akt via a dual mechanism involving NF-kappaB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTEN. Cancer Res. 2009, 69, 4107–4111. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Hui, C.; Jing, L.; Xin-Yuan, X.; Lan, S.; Zhi-Bin, Y.; Wen-Chao, L. Thymosin alpha 1 suppresses proliferation and induces apoptosis in breast cancer cells through PTEN-mediated inhibition of PI3K/Akt/mTOR signaling pathway. Apoptosis 2015, 20, 1109–1121. [Google Scholar]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, T.; Venier, R.; Dickson, B.C.; Kabaroff, L.; Alkema, M.; Chen, L.; Shern, J.F.; Yohe, M.E.; Khan, J.; Gladdy, R.A. Kras activation in p53-deficient myoblasts results in high-grade sarcoma formation with impaired myogenic differentiation. Oncotarget 2015, 6, 14220–14232. [Google Scholar] [PubMed]
- Guo, S.L.; Ye, H.; Teng, Y.; Wang, Y.L.; Yang, G.; Li, X.B.; Zhang, C.; Yang, X.; Yang, Z.Z.; Yang, X. Akt-p53-miR-365-cyclin D1/cdc25A axis contributes to gastric tumorigenesis induced by PTEN deficiecy. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M.; Llovet, J.M.; Beaugrand, M.; Lencioni, R.; Burroughs, AK.; Christensen, E.; Pagliaro, L.; Colombo, M.; Roddes, J. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J. Hepatol. 2001, 35, 421–430. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.-F.; Ou, Q.; Dai, H.; Liu, C.-A. Lentiviral-Mediated Short Hairpin RNA Knockdown of MTDHInhibits Cell Growth and Induces Apoptosis by Regulatingthe PTEN/AKT Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2015, 16, 19419-19432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819419
Li W-F, Ou Q, Dai H, Liu C-A. Lentiviral-Mediated Short Hairpin RNA Knockdown of MTDHInhibits Cell Growth and Induces Apoptosis by Regulatingthe PTEN/AKT Pathway in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2015; 16(8):19419-19432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819419
Chicago/Turabian StyleLi, Wen-Fang, Qin Ou, Hang Dai, and Chang-An Liu. 2015. "Lentiviral-Mediated Short Hairpin RNA Knockdown of MTDHInhibits Cell Growth and Induces Apoptosis by Regulatingthe PTEN/AKT Pathway in Hepatocellular Carcinoma" International Journal of Molecular Sciences 16, no. 8: 19419-19432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819419