
The Role of Autophagy in Lupus Nephritis

Abstract
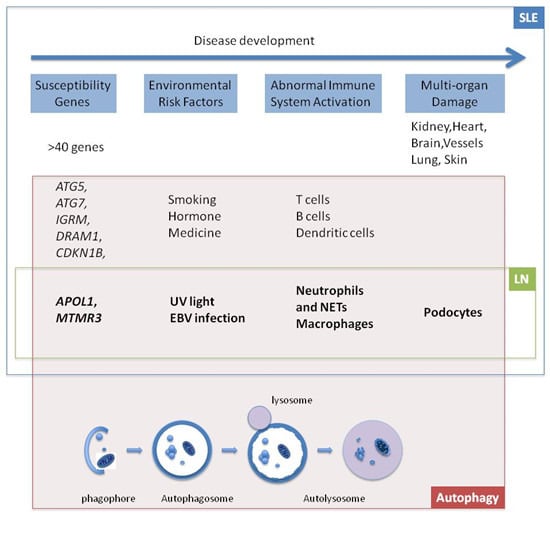
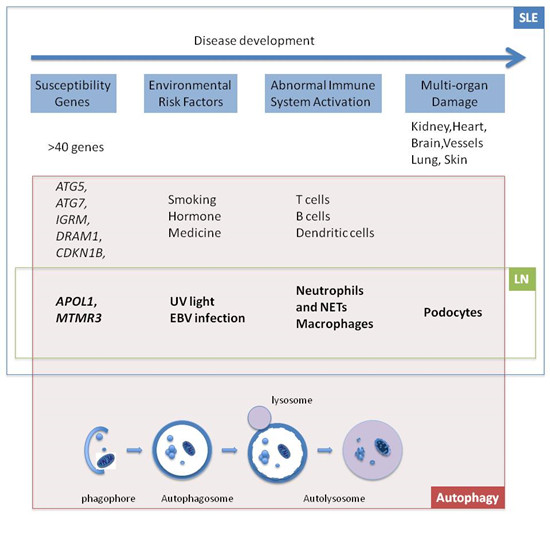
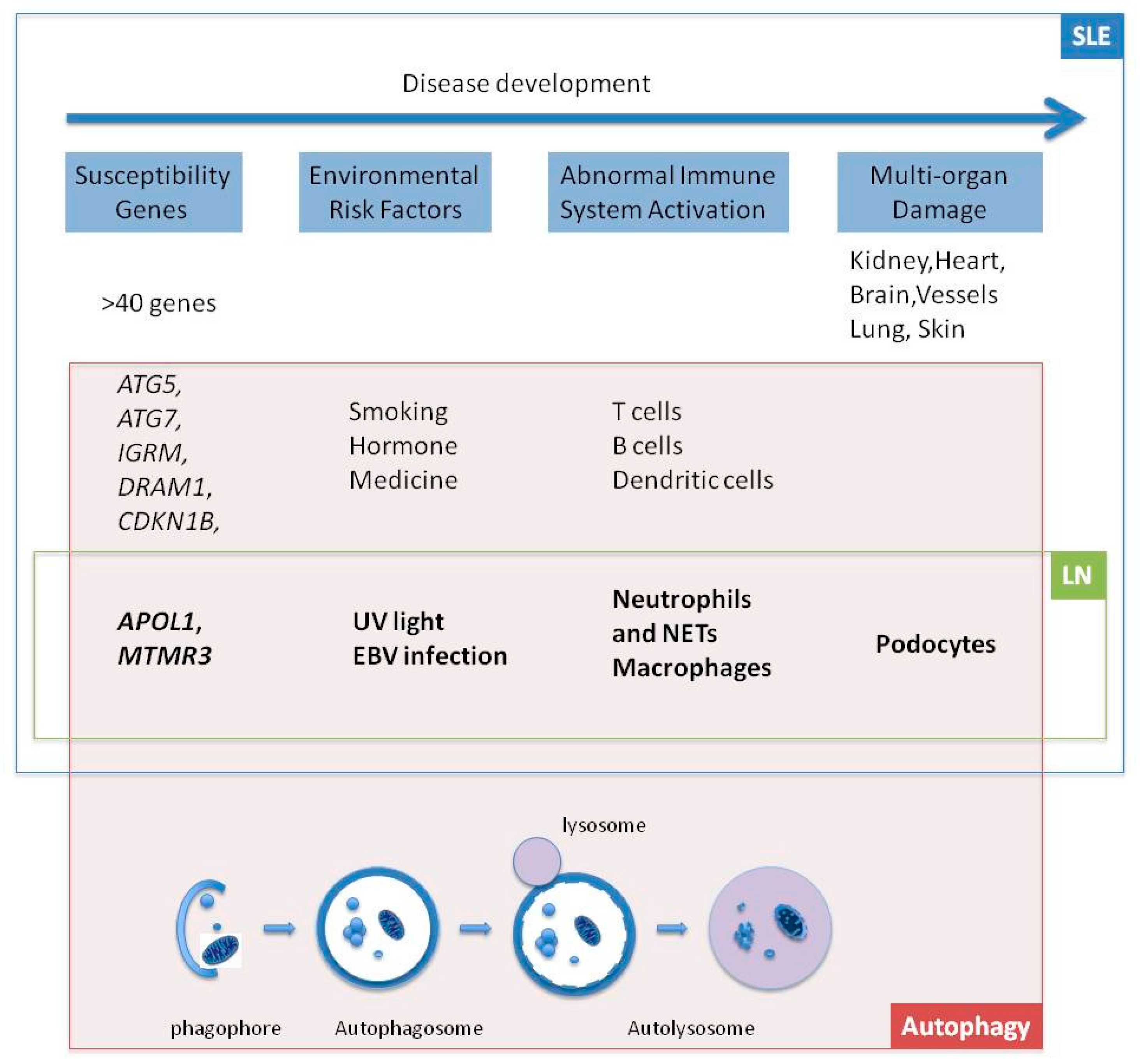
:
{kind=link}
{kind=link}
1. Introduction
2. Genes
2.1. APOL1
2.2. MTMR3
3. Environmental Risk Factors
3.1. UV Light
3.2. Epstein-Barr Virus (EBV)
4. Cells
4.1. Neutrophils and Neutrophil Extracellular Trap (NET)
4.2. Macrophages
4.3. Podocytes
5. Drugs
5.1. Inhibition of Activated mTOR Pathway in Lupus Nephritis
5.2. P140
6. Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lech, M.; Anders, H.J. The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Maroz, N.; Segal, M.S. Lupus nephritis and end-stage kidney disease. Am. J. Med. Sci. 2013, 346, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Munz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein atg16l1 enhances endotoxin-induced il-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits Il-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Zhang, H. Autophagy in immunity: Implications in etiology of autoimmune/autoinflammatory diseases. Autophagy 2012, 8, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Cheng, F.J.; Zhang, H. Emerging view of autophagy in systemic lupus erythematosus. Int. Rev. Immunol. 2015, 34, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- International Consortium for Systemic Lupus Erythematosus Group; Harley, J.B.; Alarcon-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Lu, X.L.; Lv, J.C.; Yang, H.Z.; Qin, L.X.; Zhao, M.H.; Su, Y.; Li, Z.G.; Zhang, H. Genetic association of PRDM1-Atg5 intergenic region and autophagy with systemic lupus erythematosus in a chinese population. Ann. Rheum. Dis. 2011, 70, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tang, H.; Zhang, Y.; Tang, X.; Zhang, J.; Sun, L.; Yang, J.; Cui, Y.; Zhang, L.; Hirankarn, N.; et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in asians. Am. J. Hum. Genet. 2013, 92, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.I.; Langefeld, C.D.; Andringa, K.K.; Croker, J.A.; Williams, A.H.; Garner, N.E.; Birmingham, D.J.; Hebert, L.A.; Hicks, P.J.; Segal, M.S.; et al. End-stage renal disease in african americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014, 66, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Nath, S.K.; Qi, Y.Y.; Cheng, F.J.; Yang, H.Z.; Zhang, Y.; Yang, W.; Ma, J.Y.; Zhao, M.H.; Shen, N.; et al. Brief report: Identification of MTMR3 as a novel susceptibility gene for lupus nephritis in northern han chinese by shared-gene analysis with IgA nephropathy. Arthritis Rheumatol. 2014, 66, 2842–2848. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Zhaorigetu, S.; Liu, Z.; Kaini, R.; Jiang, Z.; Hu, C.A. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 2008, 283, 21540–21549. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Beggs, M.L.; Saeed, M.; Walker, P.D. Apolipoprotein l1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J. Am. Soc. Nephrol. 2013, 24, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Winkler, C.A.; Zhao, X.; Radeva, M.K.; Gassman, J.J.; D’Agati, V.D.; Nast, C.C.; Wei, C.; Reiser, J.; Guay-Woodford, L.M.; et al. Clinical features and histology of apolipoprotein L1-associated nephropathy in the FSGS clinical trial. J. Am. Soc. Nephrol. 2015, 26, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Freedman, B.I. Apolipoprotein L1-associated nephropathy and the future of renal diagnostics. J. Am. Soc. Nephrol. 2015, 26, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Vergne, I.; Deretic, V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010, 584, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Taguchi-Atarashi, N.; Hamasaki, M.; Matsunaga, K.; Omori, H.; Ktistakis, N.T.; Yoshimori, T.; Noda, T. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 2010, 11, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Bijl, M.; Kallenberg, C.G. Ultraviolet light and cutaneous lupus. Lupus 2006, 15, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 370516. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.S.; Rothfield, N.F.; Niederman, J.C. Raised antibody titres to EB virus in systemic lupus erythematosus. Lancet 1971, 1, 167–168. [Google Scholar] [CrossRef]
- Simard, J.F.; Costenbader, K.H.; Liang, M.H.; Karlson, E.W.; Mittleman, M.A. Exposure to maternal smoking and incident SLE in a prospective cohort study. Lupus 2009, 18, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Tony, H.P.; Brocker, E.B.; Kneitz, C. Sun-induced life-threatening Lupus Nephritis. Ann. N. Y. Acad. Sci. 2007, 1108, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. UV light potentiates STING (stimulator of interferon genes)-dependent innate immune signaling through deregulation of ULK1 (Unc51-like kinase 1). J. Biol. Chem. 2015, 290, 12184–12194. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Templeton, A.K.; Guthridge, J.M.; Brown, E.J.; Harley, J.B.; James, J.A. Aberrant epstein-barr viral infection in systemic lupus erythematosus. Autoimmun. Rev. 2009, 8, 337–342. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Neas, B.R.; Moser, K.L.; Hall, T.; Bruner, G.R.; Sestak, A.L.; Harley, J.B. Systemic lupus erythematosus in adults is associated with previous epstein-barr virus exposure. Arthritis Rheum. 2001, 44, 1122–1126. [Google Scholar] [CrossRef]
- Harley, J.B.; James, J.A. Epstein-barr virus infection may be an environmental risk factor for systemic lupus erythematosus in children and teenagers. Arthritis Rheum. 1999, 42, 1782–1783. [Google Scholar] [CrossRef]
- McClain, M.T.; Heinlen, L.D.; Dennis, G.J.; Roebuck, J.; Harley, J.B.; James, J.A. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 2005, 11, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Yao, C.W.; Tao, J.L.; Yang, C.; Luo, M.N.; Li, S.M.; Liu, H.F. The expression of renal epstein-barr virus markers in patients with lupus nephritis. Exp. Ther. Med. 2014, 7, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, X.; Liao, W.; Yi, Z.; Yang, H.; Xiang, W. The expression of EBV-encoded LMP1 in young patients with lupus nephritis. Int. J. Clin. Exp. Med. 2015, 8, 6073–6078. [Google Scholar] [PubMed]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.L.; Kuballa, P.; Khor, B.; Zhang, M.; Shi, H.N.; Virgin, H.W.; Xavier, R.J. Atg5 regulates plasma cell differentiation. Autophagy 2013, 9, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Gros, F.; Arnold, J.; Page, N.; Decossas, M.; Korganow, A.S.; Martin, T.; Muller, S. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 2012, 8, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X. Systemic lupus erythematosus and the neutrophil. N. Engl. J. Med. 2011, 365, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Valle, F.; Balada, E.; Ordi-Ros, J.; Bujan-Rivas, S.; Sellas-Fernandez, A.; Vilardell-Tarres, M. Dnase 1 activity in patients with systemic lupus erythematosus: Relationship with epidemiological, clinical, immunological and therapeutical features. Lupus 2009, 18, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Itakura, A.; McCarty, O.J. Pivotal role for the mtor pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am. J. Physiol. Cell Physiol. 2013, 305, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, P.Y.; Reeves, W.H. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch. Immunol. Ther. Exp. 2010, 58, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yue, Y.; Dong, C.; Shi, Y.; Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 2014, 32, 705–714. [Google Scholar] [PubMed]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. Trehalose, an mtor independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS ONE 2014, 9, e113520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, O.; Jasiek, M.; Henique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015. [Google Scholar] [CrossRef] [PubMed]
- Anja Hillmann, H.W.; Pap, T.; Jacobi, A. Uptake of sle autoantibodies by podocytes. Ann. Rheum. Dis. 2012, 71, A1–A93. [Google Scholar] [CrossRef]
- Chan, T.M. Treatment of severe lupus nephritis: The new horizon. Nat. Rev. Nephrol. 2015, 11, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Harr, M.W.; McColl, K.S.; Zhong, F.; Molitoris, J.K.; Distelhorst, C.W. Glucocorticoids downregulate Fyn and inhibit IP3-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy 2010, 6, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 2015, 1346, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Legault, H.M.; Sypek, J.P.; Collins, M.J.; Goad, E.; Goldman, S.J.; Liu, W.; Murray, S.; Dorner, A.J.; O’Toole, M. Mapping similarities in mtor pathway perturbations in mouse lupus nephritis models and human lupus nephritis. Arthritis Res. Ther. 2008, 10, R127. [Google Scholar] [CrossRef] [PubMed]
- Lui, S.L.; Tsang, R.; Chan, K.W.; Zhang, F.; Tam, S.; Yung, S.; Chan, T.M. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol. Dial. Transplant. 2008, 23, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.; Ma, M.K.; Tang, C.S.; Chan, T.M. Proliferation signal inhibitors in the treatment of lupus nephritis: Preliminary experience. Nephrology 2012, 17, 676–680. [Google Scholar] [PubMed]
- Stylianou, K.; Petrakis, I.; Mavroeidi, V.; Stratakis, S.; Vardaki, E.; Perakis, K.; Stratigis, S.; Passam, A.; Papadogiorgaki, E.; Giannakakis, K.; et al. The PI3K/Akt/mTOR pathway is activated in murine lupus nephritis and downregulated by rapamycin. Nephrol. Dial. Transplant. 2011, 26, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Marti, H.P.; Frey, F.J. Nephrotoxicity of rapamycin: An emerging problem in clinical medicine. Nephrol. Dial. Transplant. 2005, 20, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, X.; Duan, J.; Hinrichs, D.; Wegmann, K.; Zhang, G.L.; Hall, M.; Rosenbaum, J.T. Low dose rapamycin exacerbates autoimmune experimental uveitis. PLoS ONE 2012, 7, e36589. [Google Scholar] [PubMed]
- Schall, N.; Muller, S. Resetting the autoreactive immune system with a therapeutic peptide in lupus. Lupus 2015, 24, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Monneaux, F.; Lozano, J.M.; Patarroyo, M.E.; Briand, J.P.; Muller, S. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MRL/Ipr mice. Eur. J. Immunol. 2003, 33, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Page, N.; Gros, F.; Schall, N.; Decossas, M.; Bagnard, D.; Briand, J.P.; Muller, S. Hsc70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Law, H.K.W. The Role of Autophagy in Lupus Nephritis. Int. J. Mol. Sci. 2015, 16, 25154-25167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025154
Wang L, Law HKW. The Role of Autophagy in Lupus Nephritis. International Journal of Molecular Sciences. 2015; 16(10):25154-25167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025154
Chicago/Turabian StyleWang, Linlin, and Helen Ka Wai Law. 2015. "The Role of Autophagy in Lupus Nephritis" International Journal of Molecular Sciences 16, no. 10: 25154-25167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025154