
TP53inp1 Gene Is Implicated in Early Radiation Response in Human Fibroblast Cells

 and
and
Abstract
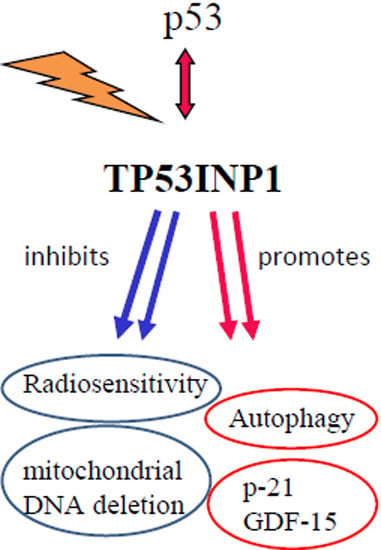
:
1. Introduction
2. Results
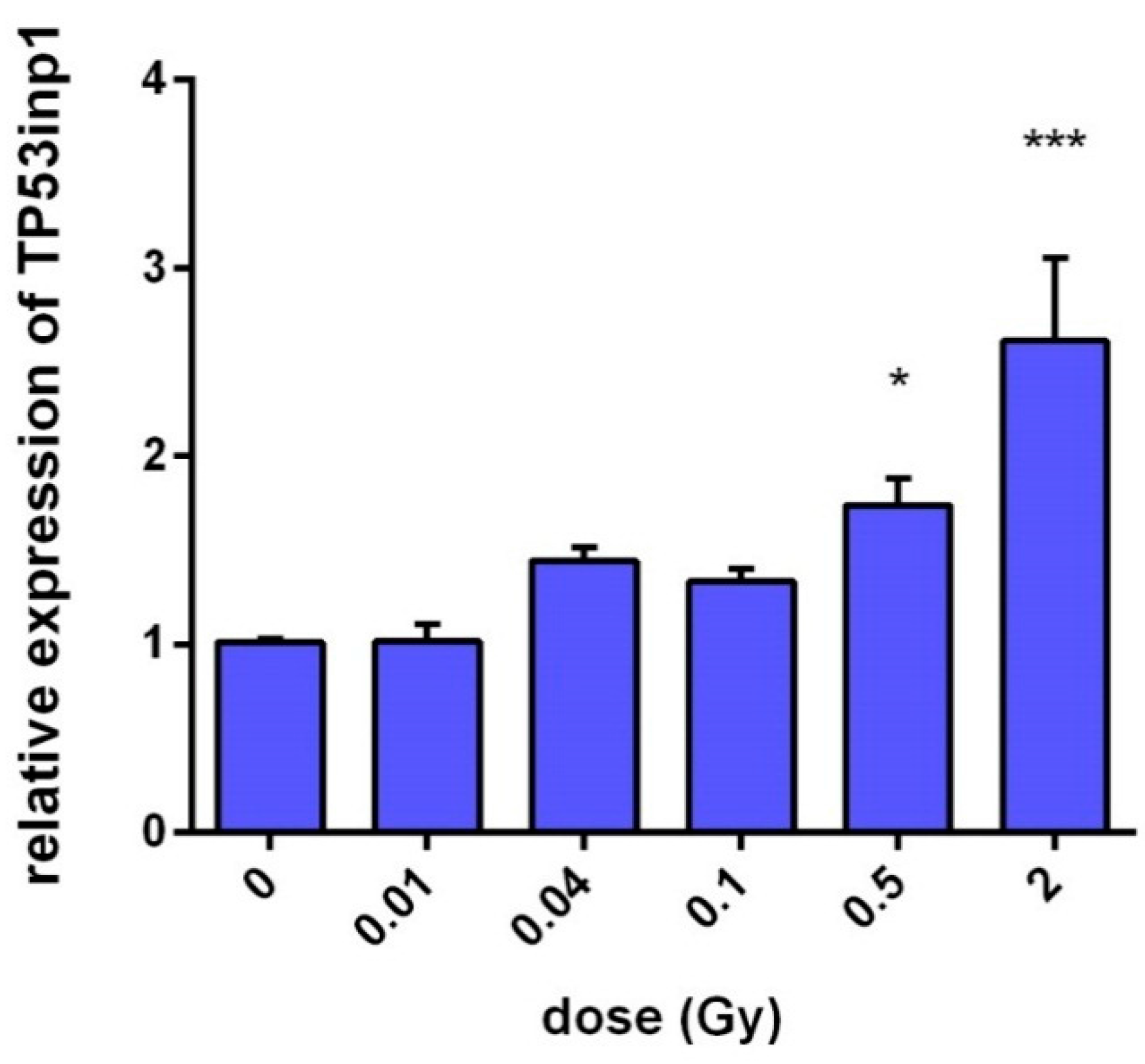
2.1. TP53inp1 Is a Radiation Response Gene in Fibroblast Cells

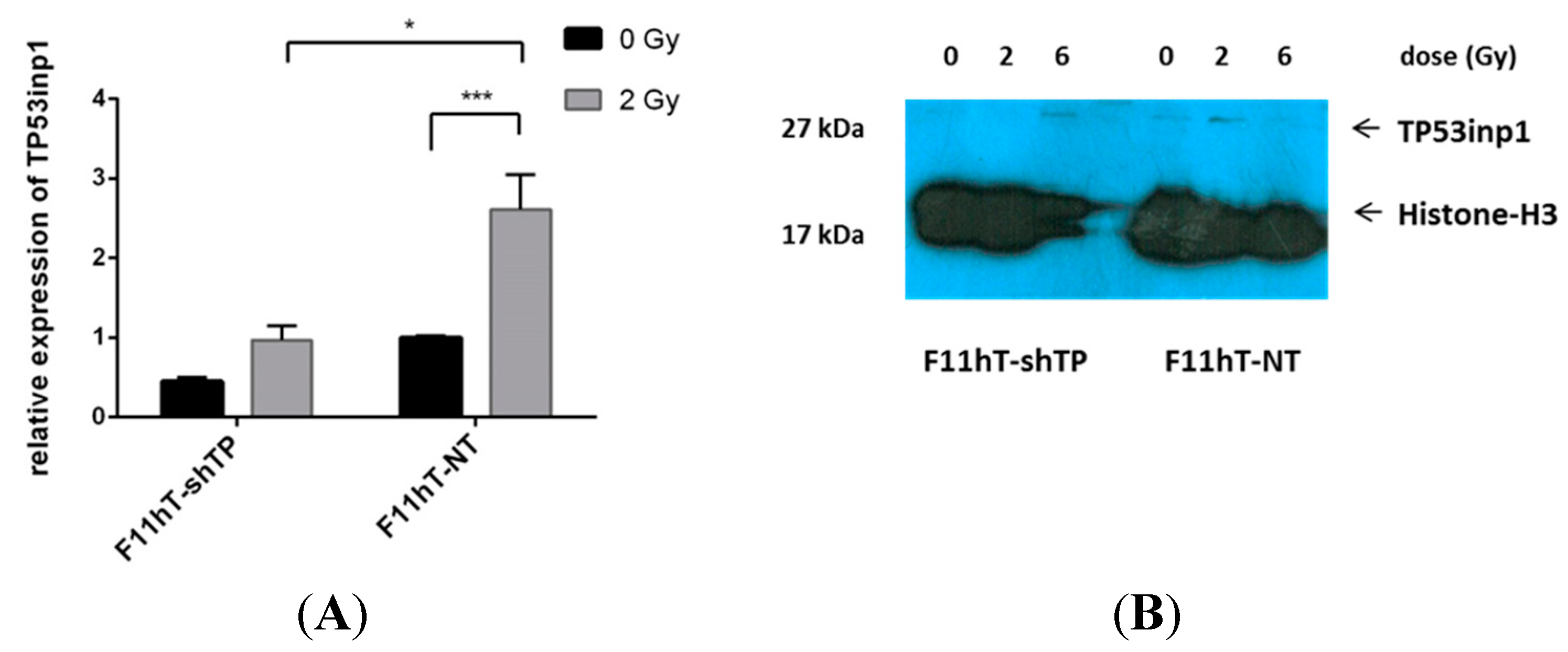
2.2. Lentiviral Delivery of TP53inp1-Targeting shRNA Effectively Decreases TP53inp1 Expression and Increases Radiation Sensitivity


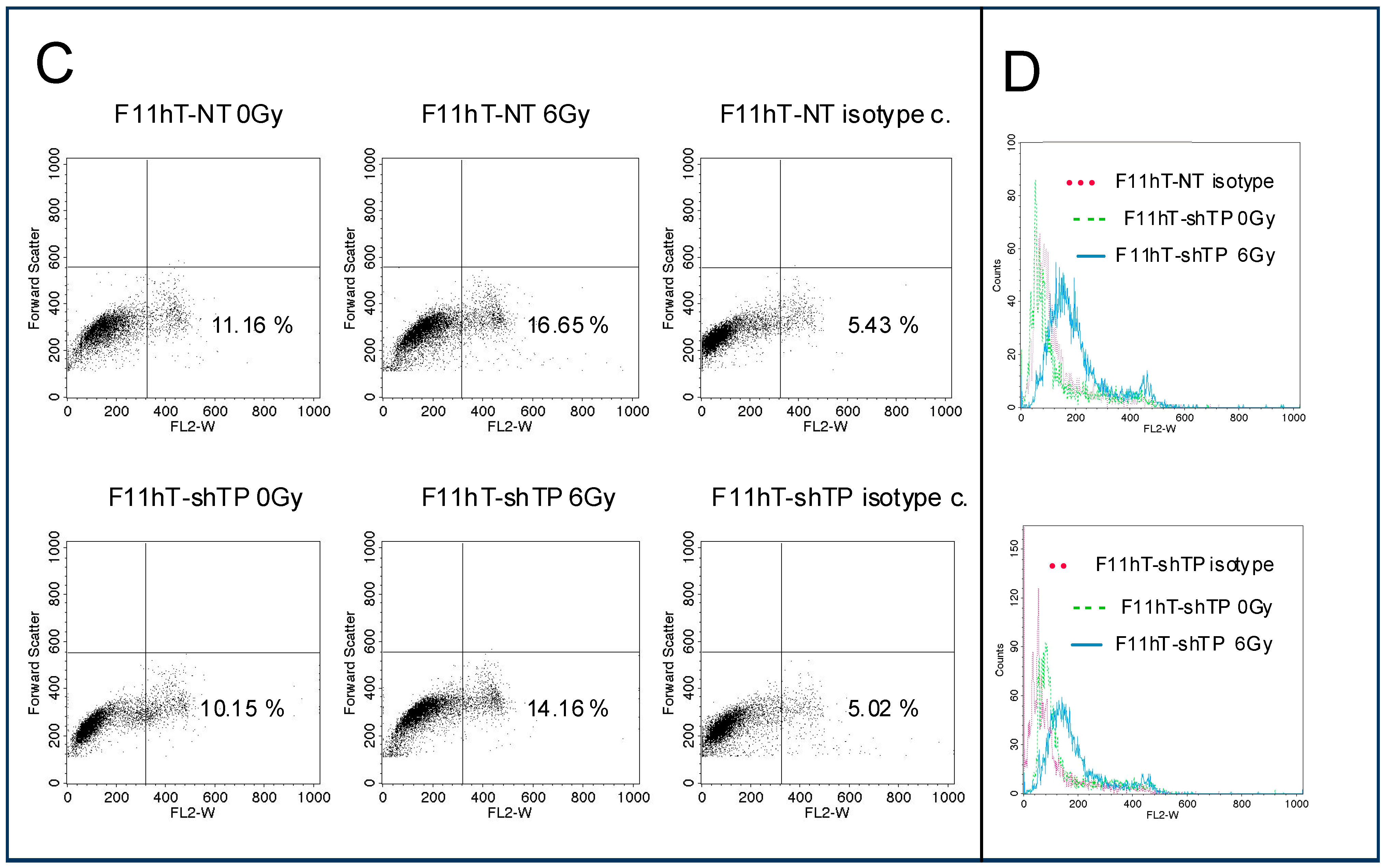
2.3. TP53inp1 Is Implicated in Autophagy


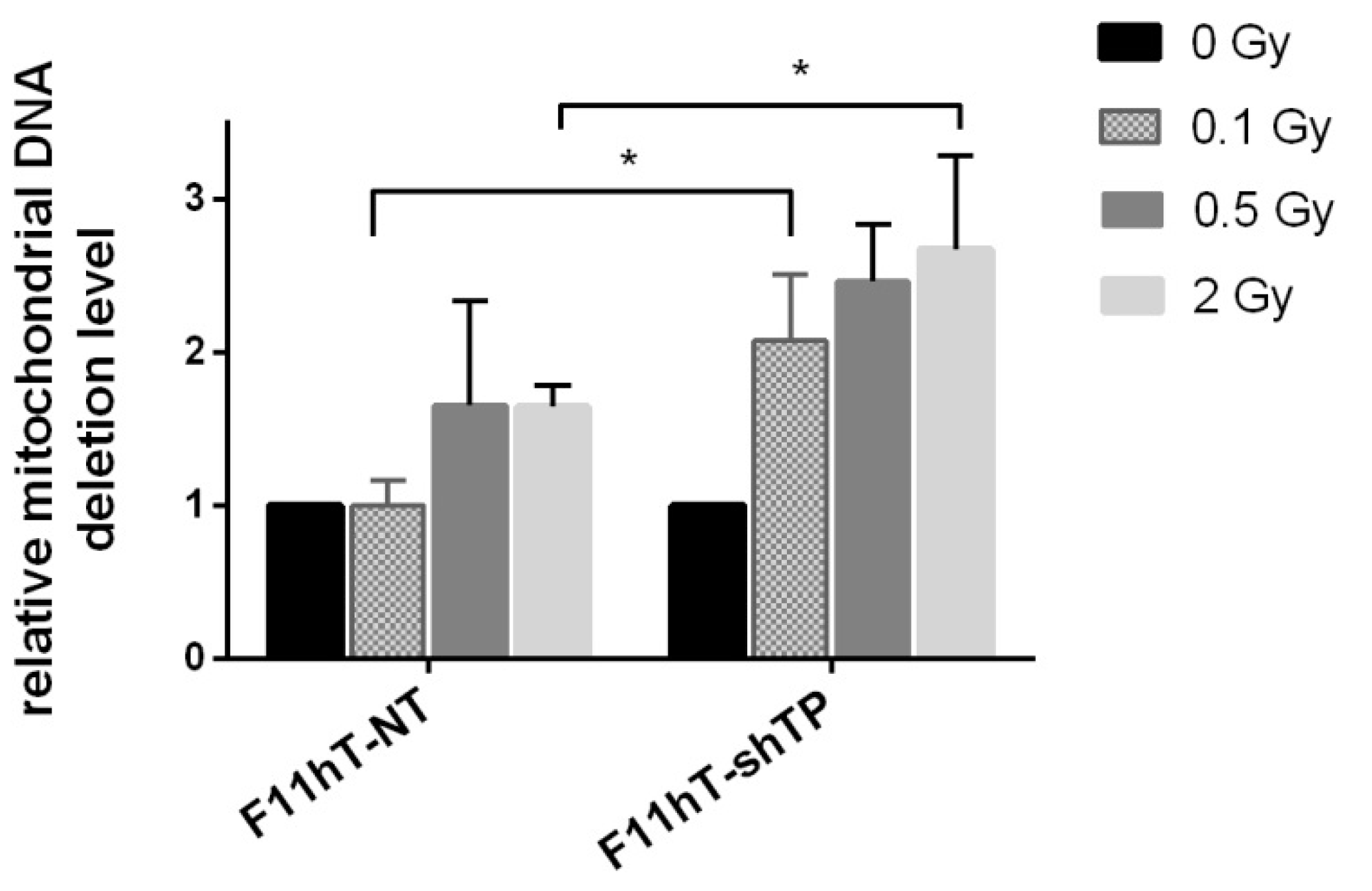
2.4. TP53inp1 Enhances the Accumulation of Common Deletion (CD) in Mitochondrial Genome


2.5. TP53inp1 Does Not Regulate IR-Induced Cellular Senescence
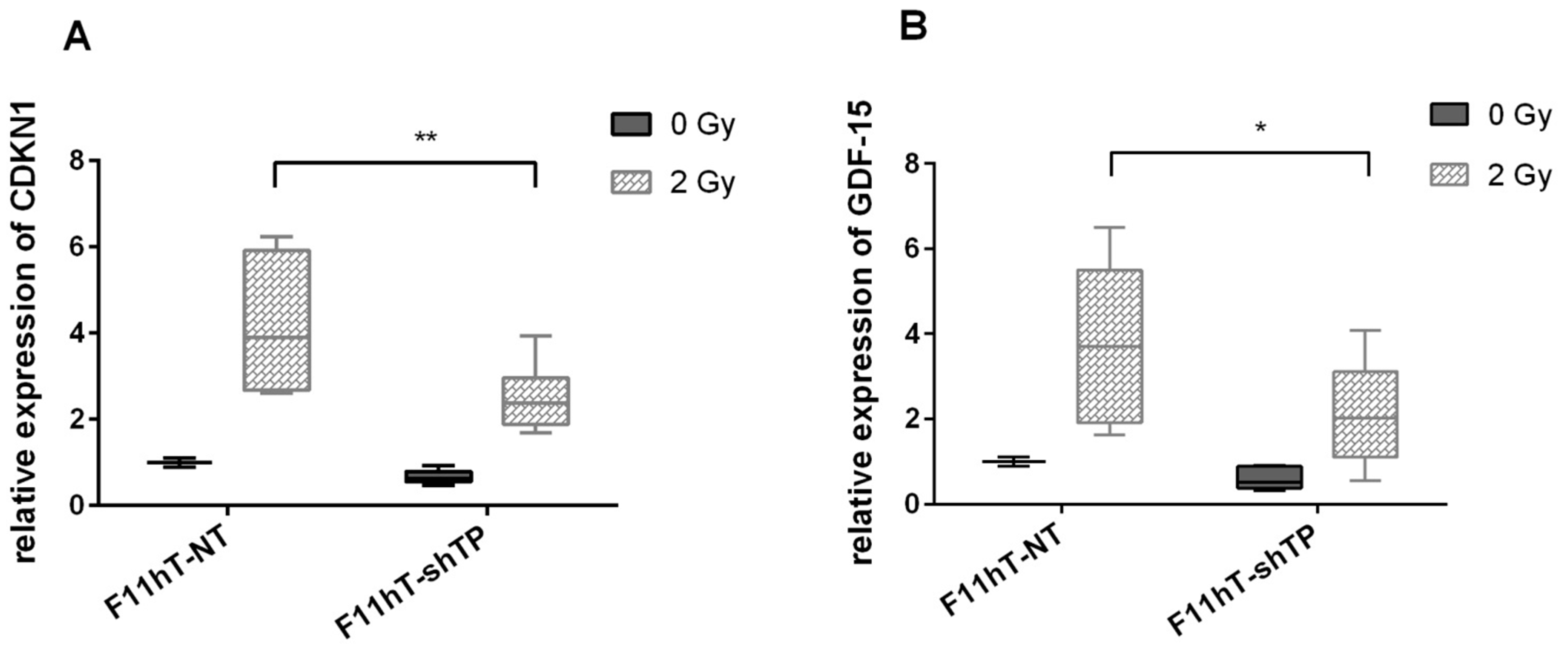
2.6. TP53inp1 Modify the Radiation-Induced Expression of CDKN1A and GDF-15 Gene

3. Discussion
4. Experimental Section
4.1. Cell Lines
4.2. Establishment of Stable shTP53inp1 Expressing Cell Lines
4.3. Radiation Treatment, Colony Forming Assay
4.4. RNA Isolation and Real-Time Quantitative PCR (qPCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| TP53INP1 | TCAGCAGAAGAAGAAGAAGAAGAG | AGCAGGAATCACTTGTATCAGC |
| CDKN1A | CCTCATCCCGTGTTCTCCTTT | GTACCACCCAGCGGACAAGT |
| GDF-15 | TCACGCCAGAAGTGCGGCTG | CGTCCCACGACCTTGACGCC |
| GAPDH | CGACCACTTTGTCAAGCTCA | AGGGGTCTACATGGCAACTG |
| ACTIN | TTGCCGACAGGATGCAGAAGGA | AGGTGGACAGCGAGGCCAGGAT |
| mtDel | CCCACTGTAAAGCTAACTTAGCATTAACC | GGTTTCGATGATGAGGTCTTTG |
| mtWT | CTGAGCCTTTTACCACTCCAG | GGTGATTGATACTCCTGATGCG |
4.5. Detection of Autophagic Vacuoles by Acridine Orange
4.6. Western Blotting
4.7. Flow Cytometry ANALYSIS of LC3B
4.8. Senescence-Associated β-Galactosidase Staining
4.9. Measurement of Mitochondrial DNA Deletion (CD) by qPCR
4.10. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Okamura, S.; Arakawa, H.; Tanaka, T.; Nakanishi, H.; Ng, C.C.; Taya, Y.; Monden, M.; Nakamura, Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell 2001, 8, 85–94. [Google Scholar] [CrossRef]
- Tomasini, R.; Seux, M.; Nowak, J.; Bontemps, C.; Carrier, A.; Dagorn, J.C.; Pebusque, M.J.; Iovanna, J.L.; Dusetti, N.J. TP53INP1 is a novel p73 target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional activity. Oncogene 2005, 24, 8093–8104. [Google Scholar] [CrossRef] [PubMed]
- Sancho, A.; Duran, J.; García-España, A.; Mauvezin, C.; Alemu, E.A.; Lamark, T.; Macias, M.J.; DeSalle, R.; Royo, M.; Sala, D.; et al. DOR/Tp53inp2 and TP53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS ONE 2012, 7, e34034. [Google Scholar] [CrossRef] [PubMed]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.; Dusetti, N.J. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, R.; Samir, A.A.; Carrier, A.; Isnardon, D.; Cecchinelli, B.; Soddu, S.; Malissen, B.; Dagorn, J.C.; Iovanna, J.L.; Dusetti, N.J. TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J. Biol. Chem. 2003, 278, 37722–37729. [Google Scholar] [CrossRef] [PubMed]
- Rieger, K.E.; Chu, G. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res. 2004, 32, 4786–4803. [Google Scholar] [CrossRef] [PubMed]
- Hegyesi, H.; Sándor, N.; Schilling, B.; Kis, E.; Lumniczky, K.; Sáfrány, G. Differentially expressed genes associated with low-dose γ radiation: Growth Differentiation Factor (GDF-15) as a radiation response gene and radiosensitizing target. In Radiation Damage in Biomolecular Systems; Garcia, G., Fuss, M.C., Eds.; Springer: New York City, NY, USA, 2012; pp. 359–370. [Google Scholar]
- Sokolov, M.V.; Neumann, R.D. Human embryonic stem cell responses to ionizing radiation exposures: current state of knowledge and future challenges. Stem Cell. Int. 2012, 2012, 579104. [Google Scholar] [CrossRef] [PubMed]
- Kis, E.; Szatmari, T.; Keszei, M.; Farkas, R.; Esik, O.; Lumniczky, K.; Falus, A.; Safrany, G. Microarray analysis of radiation response genes in primary human fibroblasts. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Lock, R.; Liu, T. Tumor Protein 53-Induced Nuclear Protein 1 Enhances p53 Function and Represses Tumorigenesis. Front Genet. 2013, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H.; Taxman, D.J. Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed]
- Panchal, N.; Chikte, S.; Wilbourn, B.R.; Meier, U.C.; Warnes, G. Flow cytometric measurement of cell organelle autophagy. In Autophagy—A Double-Edged Sword—Cell Survival or Death? Bailly, Y., Ed.; InTech: Rijeka, Croatia, 2013; pp. 65–78. [Google Scholar]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Gommeaux, J.; Pietri, S.; Culcasi, M.; Garcia, S.; Seux, M.; Barelier, S.; Vasseur, S.; Spoto, R.P.; Pebusque, M.J.; et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res. 2009, 69, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, R.; Samir, A.A.; Pebusque, M.J.; Calvo, E.L.; Totaro, S.; Dagorn, J.C.; Dusetti, N.J.; Iovanna, J.L. P53-dependent expression of the stress-induced protein (SIP). Eur. J. Cell Biol. 2002, 81, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.H.; Motoo, Y.; Garcia, S.; Iovanna, J.L.; Pebusque, M.J.; Sawabu, N. Down-expression of tumor protein p53-induced nuclear protein 1 in human gastric cancer. World J. Gastroenterol. 2006, 12, 691–696. [Google Scholar] [PubMed]
- Gironella, M.; Seux, M.; Xie, M.J.; Cano, C.; Tomasini, R.; Gommeaux, J.; Garcia, S.; Nowak, J.; Yeung, M.L.; Jeang, K.T.; et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc. Natl. Acad. Sci. USA 2007, 104, 16170–16175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gommeaux, J.; Cano, C.; Garcia, S.; Gironella, M.; Pietri, S.; Culcasi, M.; Pebusque, M.J.; Malissen, B.; Dusetti, N.J.; Iovanna, J.; et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Mol. Cell. Biol. 2007, 27, 2215–2228. [Google Scholar] [CrossRef] [PubMed]
- Giusiano, S.; Garcia, S.; Andrieu, C.; Dusetti, N.J.; Bastide, C.; Gleave, M.; Taranger-Charpin, C.; Iovanna, J.L.; Rocchi, P. TP53INP1 overexpression in prostate cancer correlates with poor prognostic factors and is predictive of biological cancer relapse. Prostate 2012, 72, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Yasunaga, J.; Bennasser, Y.; Dusetti, N.J.; Masao, M.; Jeang, K.T. Role of miRNAs (miR-93 and miR-130b) in Human T-cell Leukemia Virus-1 (HTLV-1) Transformation of Cells through Tumor Suppressor TP53INP1 Downregulation. FASEB J. 2007, 21, A1028. [Google Scholar]
- Wouters, B.G. Cell death after irradiation: How, when and why cells die. In Basic Clinical Radiobiology, 4th ed.; Joiner, M., van der Kogel, A., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 27–40. [Google Scholar]
- Noda, A.; Hirai, Y.; Hamasaki, K.; Mitani, H.; Nakamura, N.; Kodama, Y. Unrepairable DNA double-strand breaks that are generated by ionising radiation determine the fate of normal human cells. J. Cell Sci. 2012, 125, 5280–5287. [Google Scholar] [CrossRef] [PubMed]
- Geara, F.B.; Peters, LJ.; Ang, K.K.; Wike, J.L.; Sivon, S.S.; Guttenberge, R.; Callender, D.L.; Malaise, E.P.; Brock, W.A. Intrinsic radiosensitivity of normal human fibroblasts and lymphocytes after high- and low-dose-rate irradiation. Cancer Res. 1992, 52, 6348–6352. [Google Scholar] [PubMed]
- Wang, Y.; Scheiber, M.N.; Neumann, C.; Calin, G.A.; Zhou, D. MicroRNA regulation of ionizing radiation-induced premature senescence. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Christos, E.Z.; Koukourakis, M.I. Radiation-induced autophagy in normal and cancer cells: Towards novel cytoprotection and radio-sensitization policies? Autophagy 2009, 5, 442–450. [Google Scholar]
- Nam, H.Y.; Myung, M.W.; Chang, H.W.; Kim, S.J.; Seong, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovann, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012, 19, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, 1437. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Lee, S.M.; Lee, Y.J.; Li, L.J.; Lee, S.J.; Lee, J.H. Protective role of autophagy in palmitate-induced INS-1 β-cell death. Endocrinology 2009, 50, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Seillier, M.; Peuget, S.; Dusetti, N.J.; Carrier, A. Antioxidant role of p53 and of its target TP53INP1. In Antioxidant Enzyme; El-Missiry, M.A., Ed.; InTech: Rijeka, Croatia, 2012; pp. 117–138. [Google Scholar]
- Mo, N.; Lu, Y.K.; Xie, W.M.; Liu, Y.; Zhou, W.X.; Wang, H.X.; Nong, L.; Jia, Y.X.; Tan, A.H.; Chen, Y.; et al. Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol. Rep. 2014, 32, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Tai, G.; Zhang, H.; Du, J.; Chen, G.; Huang, J.; Yu, J.; Cai, J.; Liu, F. TIGAR overexpression diminishes radiosensitivity of parotid gland fibroblast cells and inhibits IR-induced cell autophagy. Int. J. Clin. Exp. Pathol. 2015, 8, 4823–4829. [Google Scholar] [PubMed]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Fei, P.; El-Deiry, W.S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Vassilev, L.T. Reduced transcriptional activity in the p53 pathway of senescent cells revealed by the MDM2 antagonist nutlin-3. Aging 2009, 1, 845–854. [Google Scholar] [PubMed]
- Sándor, N.; Walter, F.R.; Bocsik, A.; Schilling-Tóth, B.; Léner, V.; Varga, Z.; Kahán, Z.; Deli, M.A.; Sáfrány, G.; Hegyesi, H. Low dose cranial irradiation-induced cerebrovascular damage is reversible in mice. PLoS ONE 2014, 9, e112397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sándor, N.; Schilling-Tóth, B.; Kis, E.; Fodor, L.; Mucsányi, F.; Sáfrány, G.; Hegyesi, H. TP53inp1 Gene Is Implicated in Early Radiation Response in Human Fibroblast Cells. Int. J. Mol. Sci. 2015, 16, 25450-25465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025450
Sándor N, Schilling-Tóth B, Kis E, Fodor L, Mucsányi F, Sáfrány G, Hegyesi H. TP53inp1 Gene Is Implicated in Early Radiation Response in Human Fibroblast Cells. International Journal of Molecular Sciences. 2015; 16(10):25450-25465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025450
Chicago/Turabian StyleSándor, Nikolett, Boglárka Schilling-Tóth, Enikő Kis, Lili Fodor, Fruzsina Mucsányi, Géza Sáfrány, and Hargita Hegyesi. 2015. "TP53inp1 Gene Is Implicated in Early Radiation Response in Human Fibroblast Cells" International Journal of Molecular Sciences 16, no. 10: 25450-25465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161025450