
A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches

 , and
, and
Abstract
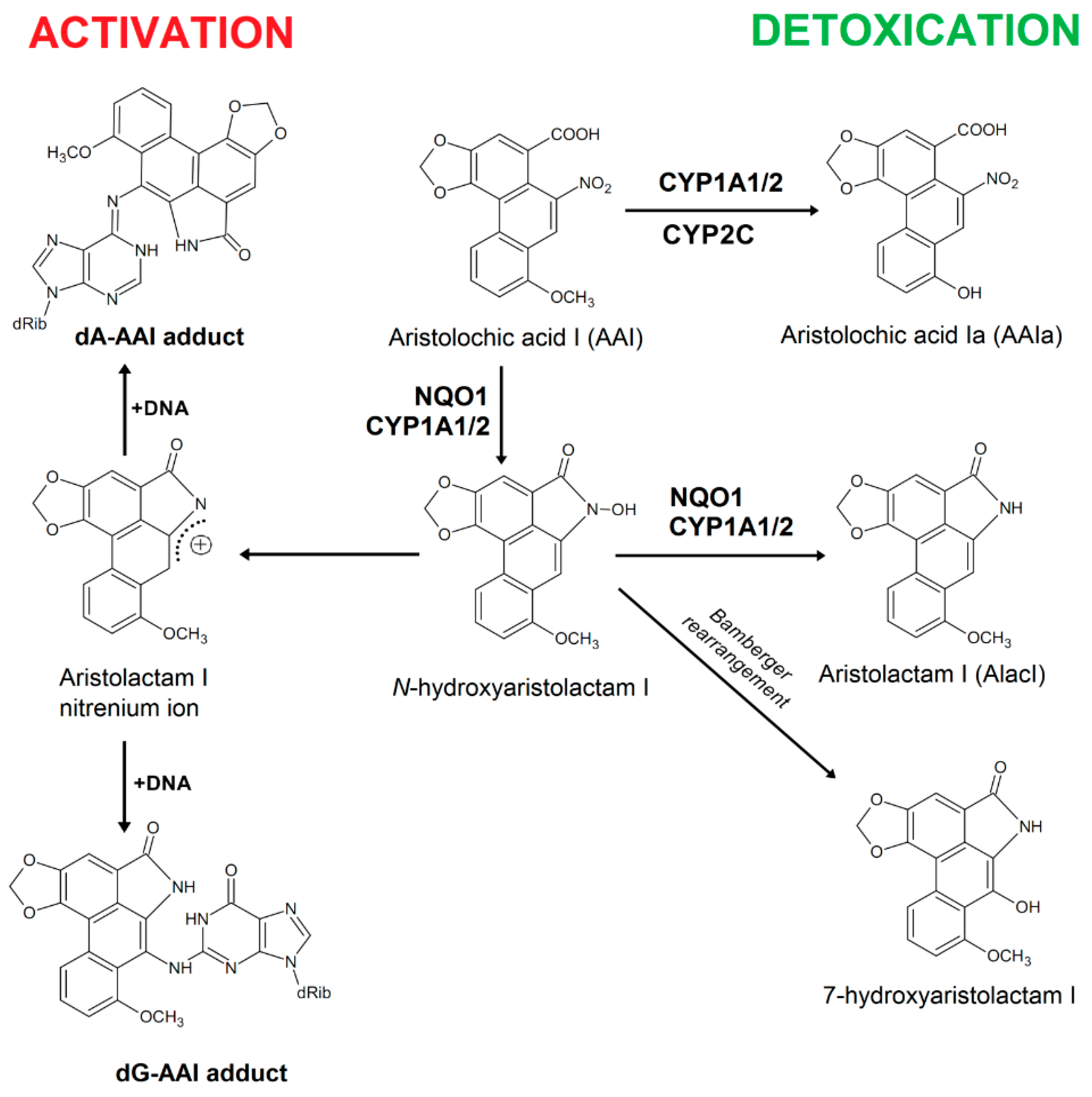
:1. Introduction

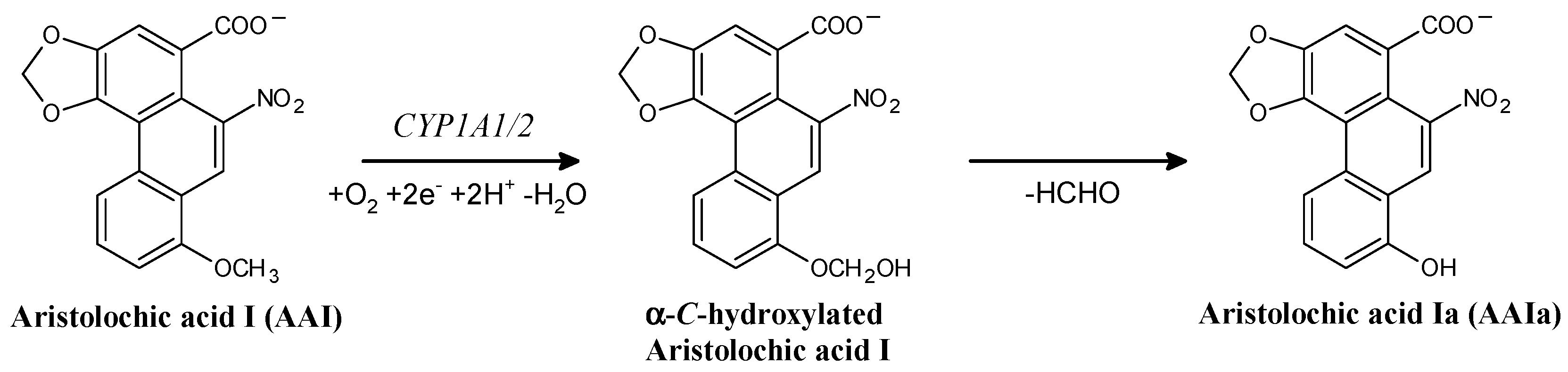
2. Results and Discussion
2.1. Effect of CYP Enzyme Inhibitors on AAI O-Demethylation Catalyzed by Human and Rat Hepatic Microsomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor a | AAIa Formation (% of Control without Inhibitor) | |
|---|---|---|
| Human Microsomes | Rat Microsomes | |
| α-Napththoflavone (CYP1A1/2) | 89 ± 5 * | 85 ± 5 ** |
| Furafylline (CYP1A2) | 75 ± 4 ** | 84 ± 5 ** |
| Diamantane (CYP2B) | NI b | NI |
| Sulfaphenazole (CYP2C) | NI | 68 ± 3 *** |
| Quinidine (CYP2D) | NI | NI |
| DDTC (CYP2A, CYP2E1) | 96 ± 5 | 52 ± 4 *** |
| Ketoconazole (CYP3A) | 74 ± 4 ** | 90 ± 4 * |
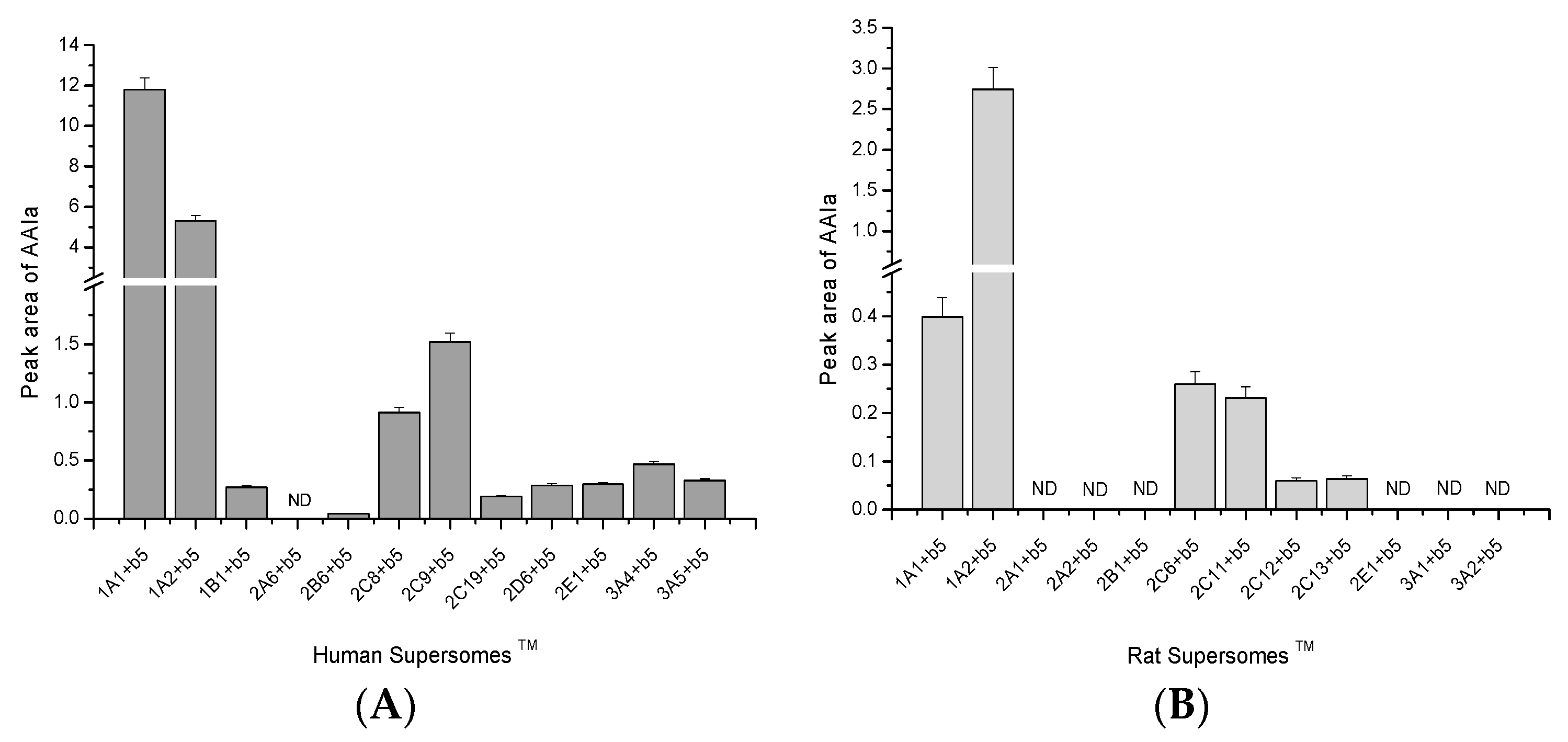
2.2. O-Demethylation of AAI to AAIa by Human and Rat Recombinant CYPs in Supersomes™

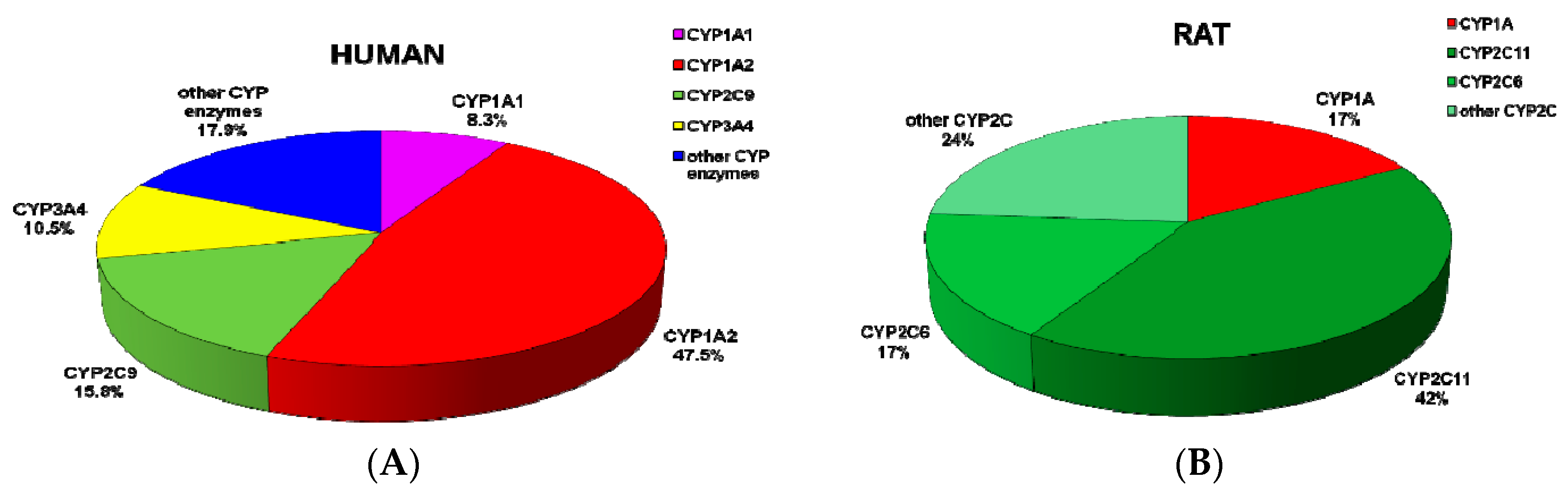
2.3. Contributions of Individual CYPs to AAIa Formation in Human and Rat Livers

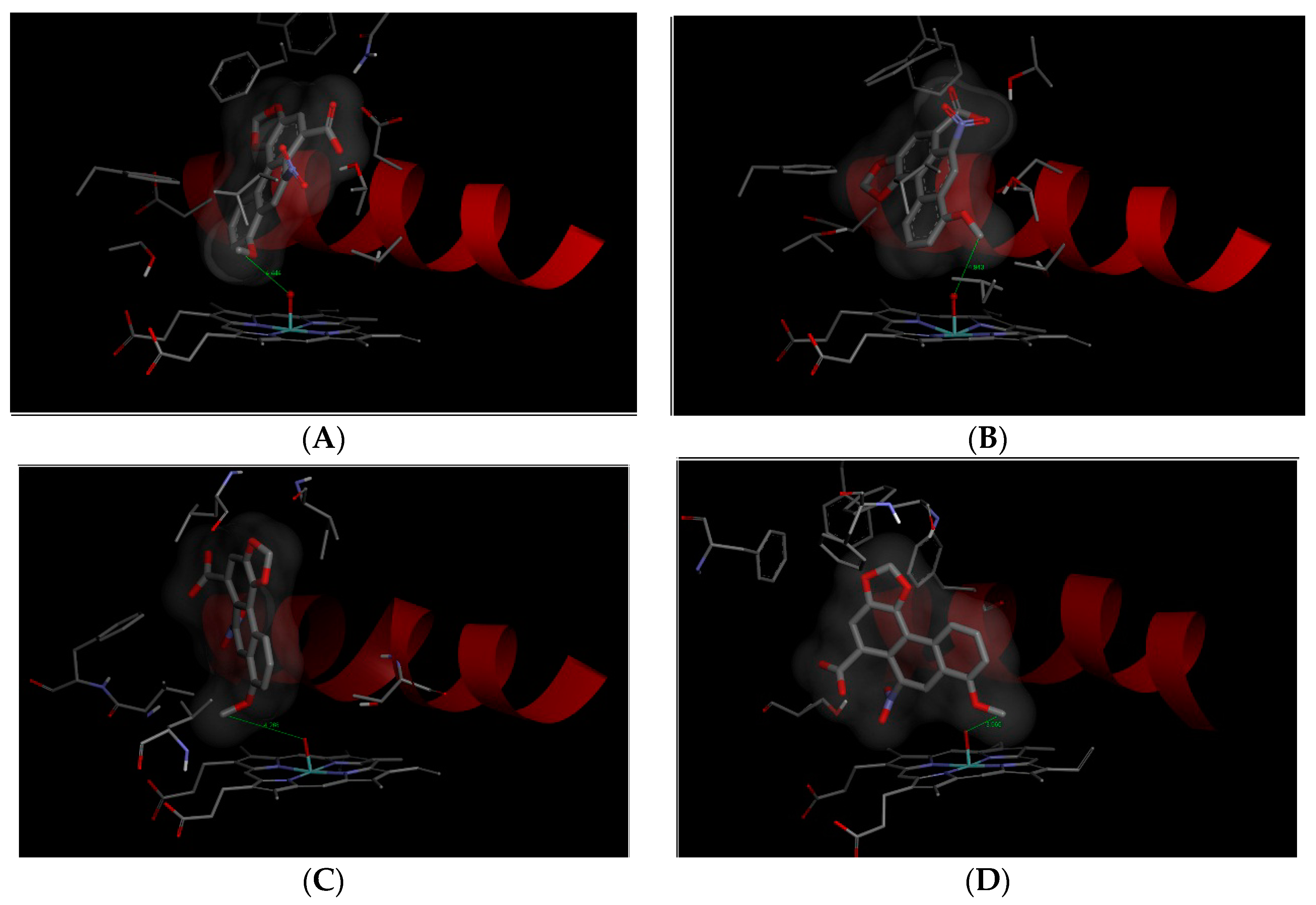
2.4. Binding of AAI to the Active Sites of Compounds I of Human CYP1A1, 1A2, 2C9, and 3A4

| Simulated System | The Most Stable Productive Orientations of AAI in the Complex with CYP | |
|---|---|---|
| Estimated Free Energy of Binding (kcal/mol) | O(Comp I)-OCH3 (AAI) Distance [Å] a | |
| CYP1A1 | −7.0 | 4.4 |
| CYP1A2 | −7.7 | 4.9 |
| CYP2C9 | −5.3 | 4.3 |
| CYP3A4 | −6.0 | 3.7 |

3. Experimental Section
3.1. Supersomes™
3.2. Preparation of Rat Hepatic Microsomes
3.3. Microsomal Incubations to Study AAI O-Demethylation
3.4. Inhibition Studies
3.5. Contributions of CYP Enzymes to O-Demethylation of AAI in Human and Rat Livers
3.6. Molecular Docking of AAI into Compounds I of Human CYP1A1, 1A2, 2C9, and 3A4
3.7. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Stiborová, M.; Arlt, V.M. Chemical and molecular basis of the carcinogenicity of Aristolochia plants. Curr. Opin. Drug Discov. Dev. 2009, 12, 141–148. [Google Scholar]
- Gökmen, M.R.; Cosyns, J.P.; Arlt, V.M.; Stiborová, M.; Phillips, D.H.; Schmeiser, H.H.; Simmonds, M.S.J.; Look, H.T.; Vanherweghem, J.L.; Nortier, J.L.; et al. The epidemiology, diagnosis and management of Aristolochic Acid Nephropathy: A narrative review. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.L.; Depierreux, M.; Tielemans, C.; Abramowicz, D.; Dratwa, M.; Jadoul, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; Vanhaelen-Fastre, R.; et al. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; de Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Rosenquist, T.A.; Sidorenko, V.; Iden, C.R.; Chen, C.H.; Pu, Y.S.; Bonala, R.; Johnson, F.; Dickman, K.G.; Grollman, A.P.; et al. Biomonitoring of aristolactam-DNA adducts in human tissues using ultra-performance liquid chromatography/ion-trap mass spectrometry. Chem. Res. Toxicol. 2012, 25, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). A review of human CARCINOGENS: Pharmaceuticals. In Environ. Health Criteria Monographs; World Health Organization: Geneva, Switzerland, 2012; Volume 100A. [Google Scholar]
- Arlt, V.M.; Stiborova, M.; vom Brocke, J.; Simoes, M.L.; Lord, G.M.; Nortier, J.L.; Hollstein, M.; Phillips, D.H.; Schmeiser, H.H. Aristolochic acid mutagenesis: Molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis 2007, 28, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeiser, H.H.; Kucab, J.E.; Arlt, V.M.; Phillips, D.H.; Hollstein, M.; Gluhovschi, G.; Gluhovschi, C.; Modilca, M.; Daminescu, L.; Petrica, L.; et al. Evidence of exposure to aristolochic acid in patients with urothelial cancer from a Balkan endemic nephropathy region of Romania. Environ. Mol. Mutagen. 2012, 53, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. Rev. Mutat. Res. 2008, 658, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Schmeiser, H.H. Biotransformation enzymes in development of renal injury and urothelial cancer caused by aristolochic acid. Kidney Int. 2008, 73, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Martínek, V.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Enzymes metabolizing aristolochic acid and their contribution to the development of Aristolochic acid nephropathy and urothelial cancer. Curr. Drug Metab. 2013, 14, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Knock-out and humanized mice as suitable tools to identify enzymes metabolizing the human carcinogen aristolochic acid. Xenobiotica 2014, 44, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele de Strihou, C.; Cosyns, J.P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar] [PubMed]
- Schmeiser, H.H.; Frei, E.; Wiessler, M.; Stiborová, M. Comparison of DNA adduct formation by aristolochic acids in various in vitro activation systems by 32P-post-labelling: Evidence for reductive activation by peroxidases. Carcinogenesis 1996, 18, 1055–1062. [Google Scholar] [CrossRef]
- Arlt, V.M.; Ferluga, D.; Stiborova, M.; Pfohl-Leszkowicz, A.; Vukelic, M.; Ceovic, S.; Schmeiser, H.H.; Cosyns, J.P. Is aristolochic acid a risk factor for Balkan endemic nephropathy-associated urothelial cancer? Int. J. Cancer 2002, 101, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Nortier, J.L.; Singh, R.; da Costa, G.G.; Sennesael, J.; Cassuto-Viguier, E.; Ambrosetti, D.; Rorive, S.; Pozdzik, A.; Phillips, D.H.; et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int. J. Cancer 2014, 135, 562–567. [Google Scholar] [CrossRef]
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 2014, 43, e11–e17. [Google Scholar] [CrossRef]
- Nedelko, T.; Arlt, V.M.; Phillips, D.H.; Hollstein, M. TP53 mutation signature supports involvement of aristolochic acid in the aetiology of endemic nephropathy-associated tumours. Int. J. Cancer 2009, 124, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Phillips, D.H.; Arlt, V.M. Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J. 2010, 277, 2567–2583. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.L.; Pang, S.T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-wide mutational signatures of aristolochic Acid and its application as a screening tool. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Schmeiser, H.H.; Straif, K.; Wild, C.P. Upper urinary tract urothelial cancer: Where it is A:T. Nat. Rev. 2012, 12, 503–504. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Cu, L.; Xu, G.; Cai, Z. Study of the phase I and phase II metabolism of nephrotoxin aristolochic acid by liquid chromatography/tandem mass spectrometry. Rapid. Commun. Mass Spectrom. 2006, 20, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Bonala, R.R.; Rosenquist, T.; Rieger, R.; Suzuki, N.; Johnson, F.; Miller, F.; Grollman, A.P. Detoxification of aristolochic acid I by O-demethylation: Less nephrotoxicity and genotoxicity of aristolochic acid Ia in rodents. Int. J. Cancer 2010, 127, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levova, K.; Barta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborova, M. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: Studies in Cyp1a1−/−, Cyp1a2−/−, and Cyp1a1/1a2−/− mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus detoxication of the urothelial carcinogen aristolochic acid I by human cytochrome P450 1A1 and 1A2. Toxicol. Sci. 2012, 125, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Wiessler, M.; Schmeiser, H.H. Carcinogenic aristolochic acids upon activation by DT-diaphorase form adducts found in DNA of patients with Chinese herbs nephropathy. Carcinogenesis 2002, 23, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Sopková, K.; Marková, V.; Laňková, M.; Kumstýřová, T.; Wiessler, M.; Schmeiser, H.H. Human cytosolic enzymes involved in the metabolic activation of carcinogenic aristolochic acid: Evidence for reductive activation by human NAD(P)H:quinone oxidoreductase. Carcinogenesis 2003, 24, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Mareš, J.; Frei, E.; Arlt, V.M.; Martínek, V.; Schmeiser, H.H. The human carcinogen aristolochic acid I is activated to form DNA adducts by human NAD(P)H:quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen. 2011, 52, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid I: Experimental and theoretical approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Šulc, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The influence of dicoumarol on the bioactivation of the carcinogen aristolochic acid I in rats. Mutagenesis 2014, 29, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Martinek, V.; Kubickova, B.; Arlt, V.M.; Frei, E.; Schmeiser, H.H.; Hudeček, J.; Stiborova, M. Comparison of activation of aristolochic acid I and II with NADPH:quinone oxidoreductase, sulphotransferases and N-acetyltranferases. Neuro Endocrinol. Lett. 2011, 32, 57–70. [Google Scholar] [PubMed]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X.; et al. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci. 2011, 122, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Wiessler, M.; Schmeiser, H.H. Human enzymes involved in the metabolic activation of carcinogenic aristolochic acids: Evidence for reductive activation by cytochromes P450 1A1 and 1A2. Chem. Res. Toxicol. 2001, 14, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human hepatic and renal microsomes, cytochromes P450 1A1/2, NADPH:CYP reductase and prostaglandin H synthase mediate the formation of aristolochic acid DNA-adducts found in patients with urothelial cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Sopko, B.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Hudeček, J. The binding of aristolochic acid I to the active site of human cytochromes P450 1A1 and 1A2 explains their potential to reductively activate this human carcinogen. Cancer Lett. 2005, 229, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Mareš, J.; Levová, K.; Pavlíčková, J.; Bárta, F.; Hodek, P.; Frei, E.; Schmeiser, H.H. Role of cytochromes P450 in metabolism of carcinogenic aristolochic acid I: Evidence of their contribution to aristolochic acid I detoxication and activation in rat liver. Neuro Endocrinol. Lett. 2011, 32, 121–130. [Google Scholar] [PubMed]
- Arlt, V.M.; Henderson, C.J.; Wolf, C.R.; Stiborova, M.; Phillips, D.H. The Hepatic Reductase Null (HRN™) and Reductase Conditional Null (RCN) mouse models as suitable tools to study metabolism, toxicity and carcinogenicity of environmental pollutants. Toxicol. Res. 2015, 4, 548–562. [Google Scholar] [CrossRef]
- Levová, K.; Mizerovská, M.; Kotrbová, V.; Šulc, M.; Henderson, C.J.; Wolf, C.R.; Philips, D.H.; Frei, E.; Schmeiser, H.H.; Mareš, J.; et al. Role of cytochromes P450 1A1/2 in detoxication and activation of carcinogenic aristolochic acid I: Studies with the hepatic NADPH: Cytochrome P450 reductase null (HRN) mouse model. Toxicol. Sci. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Levová, K.; Moserova, M.; Nebert, D.W.; Phillips, D.H.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborova, M. NAD(P)H:quinone oxidoreductase expression in Cyp1a-knockout and CYP1A-humanized mouse lines and its effect on bioactivation of the carcinogen aristolochic acid I. Toxicol. Appl. Pharmacol. 2012, 265, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Jerabek, P.; Martinek, V.; Stiborova, M. Theoretical investigation of differences in nitroreduction of aristolochic acid I by cytochromes P450 1A1, 1A2 and 1B1. Neuro Endocrinol. Lett. 2012, 33, 25–32. [Google Scholar] [PubMed]
- Sistkova, J.; Hudecek, J.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Stiborova, M. Human cytochromes P450 1A1 and 1A2 participate in detoxication of carcinogenic aristolochic acid. Neuro Endocrinol. Lett. 2008, 29, 733–737. [Google Scholar] [PubMed]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos. 2010, 38, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The influence of ochratoxin A on DNA adduct formation by the carcinogen aristolochic acid in rats. Arch. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ge, M.; Xue, X.; Wang, C.; Wang, H.; Wu, X.; Li, L.; Liu, L.; Qi, X.; Zhang, Y.; et al. Hepatic cytochrome P450s metabolize aristolochic acid and reduce its kidney toxicity. Kidney Int. 2008, 73, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Xiao, Y.; Zhu, H.; Wang, H.; Liu, Y.; Xie, T.; Ren, J. Induction of P450 1A by 3-methylcholanthrene protects mice from aristolochic acid-I-induced acute renal injury. Nephrol. Dial. Transplant. 2008, 23, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Schoepe, K.B.; Wiessler, M. DNA adduct formation of aristolochic acid I and II in vitro and in vivo. Carcinogenesis 1988, 9, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Pfau, W.; Schmeiser, H.H.; Wiessler, M. 32P-postlabelling analysis of the DNA adducts formed by aristolochic acid I and II. Carcinogenesis 1990, 11, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Fernando, R.C.; Schmeiser, H.H.; Frei, E.; Pfau, W.; Wiessler, M. Characterization of DNA adducts formed by aristolochic acids in the target organ (forestomach) of rats by 32P-postlabelling analysis using different chromatographic procedures. Carcinogenesis 1994, 15, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Bieler, C.A.; Stiborova, M.; Wiessler, M.; Cosyns, J.P.; van Ypersele de Strihou, C.; Schmeiser, H.H. 32P-post-labelling analysis of DNA adducts formed by aristolochic acid in tissues from patients with Chinese herbs nephropathy. Carcinogenesis 1997, 18, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Debelle, F.D.; Nortier, J.L.; de Prez, E.G.; Garbar, C.H.; Vienne, A.R.; Salmon, I.J.; Deschodt-Lanckman, M.M.; Vanherweghem, J.L. Aristolochic acids induce chronic renal failure with interstitial fibrosis in salt-depleted rats. J. Am. Soc. Nephrol. 2002, 13, 431–436. [Google Scholar] [PubMed]
- Bárta, F.; Levová, K.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. The effect of aristolochic acid I on expression of NAD(P)H:quinone oxidoreductase in mice and rats—A comparative study. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 768, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nedelcheva, V.; Gut, I. P450 in the rat and man: Methods of investigation, substrate specificities and relevance to cancer. Xenobiotica 1994, 24, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; DiCarlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–480. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Gillam, E.M.; Dong, M.S.; Johnson, W.W.; Guengerich, F.P.; Shimada, T. Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: Effects of cytochrome P450–P450 and cytochrome P450-b5 interactions. Arch. Biochem. Biophys. 1997, 342, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimada, T.; Martin, M.V.; Guengerich, F.P. Stimulation of cytochrome P450 reactions by APO-cytochrome b5: Evidence against transfer of heme from cytochrome P450 3A4 to APO-cytochrome b5 or heme oxygenase. J. Biol. Chem. 2001, 276, 30885–30891. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.D. The roles of cytochrome b5 in cytochrome P450 reactions. J. Biochem. Mol. Toxicol. 2002, 16, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, J.B.; Jansson, I. The many roles of cytochrome b5. Pharmacol. Ther. 2003, 97, 139–152. [Google Scholar] [CrossRef]
- Stiborova, M.; Martinek, V.; Schmeiser, H.H.; Frei, E. Modulation of CYP1A1-mediated oxidation of carcinogenic azo dye Sudan I and its binding to DNA by cytochrome b5. Neuro Endocrinol. Lett. 2006, 27, 35–39. [Google Scholar] [PubMed]
- Stiborova, M.; Indra, R.; Moserova, M.; Cerna, V.; Rupertova, M.; Martinek, V.; Eckschlager, T.; Kizek, R.; Frei, E. Cytochrome b5 increases cytochrome P450 3A4-mediated activation of anticancer drug ellipticine to 13-hydroxyellipticine whose covalent binding to DNA is elevated by sulfotransferases and N,O-acetyltransferases. Chem. Res. Toxicol. 2012, 25, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Poljaková, J.; Martínková, E.; Ulrichová, J.; Simánek, V.; Dvořák, Z.; Frei, E. Ellipticine oxidation and DNA adduct formation in human hepatocytes is catalyzed by human cytochromes P450 and enhanced by cytochrome b5. Toxicology 2012, 302, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; McLaughlin, L.A.; Ronseaux, S.; Rosewell, I.; Houston, J.B.; Henderson, C.J.; Wolf, C.R. Defining the in vivo role for cytochrome b5 in cytochrome P450 function through the conditional hepatic deletion of microsomal cytochrome b5. J. Biol. Chem. 2008, 283, 31385–31393. [Google Scholar] [CrossRef] [PubMed]
- Kotrbova, V.; Aimova, D.; Ingr, M.; Borek-Dohalska, L.; Martinek, V.; Stiborova, M. Preparation of a biologically active APO-cytochrome b5 via heterologous expression in Escherichia coli. Protein Exp. Purif. 2009, 66, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kotrbova, V.; Mrazova, B.; Moserova, M.; Martinek, V.; Hodek, P.; Hudecek, J.; Frei, E.; Stiborova, M. Cytochrome b5 shifts oxidation of the anticancer drug ellipticine by cytochromes P450 1A1 and 1A2 from its detoxication to activation, thereby modulating its pharmacological efficacy. Biochem. Pharmacol. 2011, 82, 669–680. [Google Scholar] [CrossRef] [PubMed]
- McLaughin, L.A.; Ronseaux, S.; Finn, R.D.; Henderson, C.L.; Wolf, C.R. Deletion of microsomal cytochrome b5 profoundly affects hepatic and extrahepatic drug metabolism. Mol. Pharmacol. 2010, 75, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Sulc, M.; Jecmen, T.; Snajdrova, R.; Novak, P.; Martinek, V.; Hodek, P.; Stiborova, M.; Hudecek, J. Mapping of interaction between cytochrome P450 2B4 and cytochrome b5: The first evidence of two mutual orientations. Neuro Endocrinol. Lett. 2012, 33, 41–47. [Google Scholar] [PubMed]
- Henderson, C.J.; McLaughlin, L.A.; Wolf, C.R. Evidence that cytochrome b5 and cytochrome b5 reductase can act as sole electron donors to the hepatic cytochrome P450 system. Mol. Pharmacol. 2013, 83, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Jeřábek, P.; Florián, J.; Stiborová, M.; Martínek, V. Flexible docking-based molecular dynamics/steered molecular dynamics calculations of protein-protein contacts in a complex of cytochrome P450 1A2 with cytochrome b5. Biochemistry 2014, 53, 6695–6705. [Google Scholar] [CrossRef] [PubMed]
- Večeřa, R.; Zachařová, A.; Orolin, J.; Strojil, J.; Skottová, N.; Anzenbacher, P. Fenofibrate-induced decrease of expression of CYP2C11 and CYP2C6 in rat. Biopharm. Drug Dispos. 2011, 32, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Zachařová, A.; Siller, M.; Spičáková, A.; Anzenbacherová, E.; Skottová, N.; Anzenbacher, P.; Večeřa, R. Rosuvastatin suppresses the liver microsomal CYP2C11 and CYP2C6 expression in male Wistar rats. Xenobiotica 2012, 42, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, K.; Jaiswal, A.K.; Owens, R.A.; Jones, J.E.; Nebert, D.W.; Kimura, S. Human CYP1A2: Sequence, gene structure, comparison with the mouse and rat orthologous gene, and differences in liver 1A2 mRNA expression. Mol. Endocrinol. 1989, 3, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Sutter, T.R.; Tang, Y.M.; Hayes, C.L.; Wo, Y.Y.; Jabs, E.W.; Li, X.; Yin, H.; Cody, C.W.; Greenlee, W.F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [Google Scholar] [PubMed]
- Shimada, T.; Hayes, C.L.; Yamazaki, H.; Amin, S.; Hecht, S.S.; Guengerich, F.P.; Sutter, T. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996, 56, 2979–2984. [Google Scholar] [PubMed]
- Hakkola, J.; Pasanen, M.; Pelkonen, O.; Hukkanen, J.; Evisalmi, S.; Anttila, S.; Rane, A.; Mäntylä, M.; Purkunen, R.; Saarikoski, S.; et al. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis 1997, 18, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Adams, D.A.; Watts, P.S.; Davies, D.S.; Boobis, A.R. Development of a comprehensive panel of antibodies against the major xenobiotic metabolising forms of cytochrome P450 in humans. Biochem. Pharmacol. 1998, 56, 377–387. [Google Scholar] [CrossRef]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Hodek, P.; Frei, E. Sudan I is a potential carcinogen for humans: Evidence for its metabolic activation and detoxication by human recombinant cytochrome P450 1A1 and liver microsomes. Cancer Res. 2002, 62, 5678–5684. [Google Scholar] [PubMed]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Koblas, T.; Hodek, P. Expression of cytochrome P450 1A1 and its contribution to oxidation of a potential human carcinogen 1-phenylazo-2-naphthol (Sudan I) in human livers. Cancer Lett. 2005, 220, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Leu, A.N.; Gonzalez, F.J.; Guengerich, F.P.; Chowdhury, A.R.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 9936–9951. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimizu, M.; Nagashima, T.; Minoshima, M.; Murayama, N. Rat cytochrome P450 2C11 in liver microsomes involved in oxidation of anesthetic agent propofol and deactivated by prior treatment with propofol. Drug Metab. Dispos. 2006, 34, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Indra, R.; Moserova, M.; Kroftova, N.; Sulc, M.; Martinkova, M.; Adam, V.; Eckschlager, T.; Kizek, R.; Arlt, V.M.; Stiborova, M. Modulation of human cytochrome P450 1A1-mediated oxidation of benzo[a]pyrene by NADPH:cytochrome P450 oxidoreductase and cytochrome b5. Neuro Endocrinol. Lett. 2014, 35, 105–113. [Google Scholar] [PubMed]
- Indra, R.; M0oserova, M.; Sulc, M.; Frei, E.; Stiborova, M. Oxidation of carcinogenic benzo[a]pyrene by human and rat cytochrome P450 1A1 and its influencing by cytochrome b5—A comparative study. Neuro Endocrinol. Lett. 2014, 34, 55–63. [Google Scholar]
- Stiborová, M.; Asfaw, B.; Anzenbacher, P.; Hodek, P. A new way to carcinogenicity of azo dyes: The benzenediazonium ion formed from a non-aminoazo dye, 1-phenylazo-2-hydroxynaphthalene (Sudan I) by microsomal enzymes binds to deoxyguanosine residues of DNA. Cancer Lett. 1988, 40, 327–333. [Google Scholar] [CrossRef]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Pontikis, G.; Borden, J.; Martínek, V.; Florián, J. Linear energy relationships for the octahedral preference of Mg, Ca and transition metal ions. J. Phys. Chem. 2009, 113, 3588–3593. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Robb, M.A.; Cheeseman, J.R. Gaussian 03; Gaussian Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches. Int. J. Mol. Sci. 2015, 16, 27561-27575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126047
Stiborová M, Bárta F, Levová K, Hodek P, Schmeiser HH, Arlt VM, Martínek V. A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches. International Journal of Molecular Sciences. 2015; 16(11):27561-27575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126047
Chicago/Turabian StyleStiborová, Marie, František Bárta, Kateřina Levová, Petr Hodek, Heinz H. Schmeiser, Volker M. Arlt, and Václav Martínek. 2015. "A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches" International Journal of Molecular Sciences 16, no. 11: 27561-27575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126047