
Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research

{kind=link}
Abstract
:1. Introduction
2. G Protein-Coupled Receptors
3. Epidermal Growth Factor Receptor (EGFR)
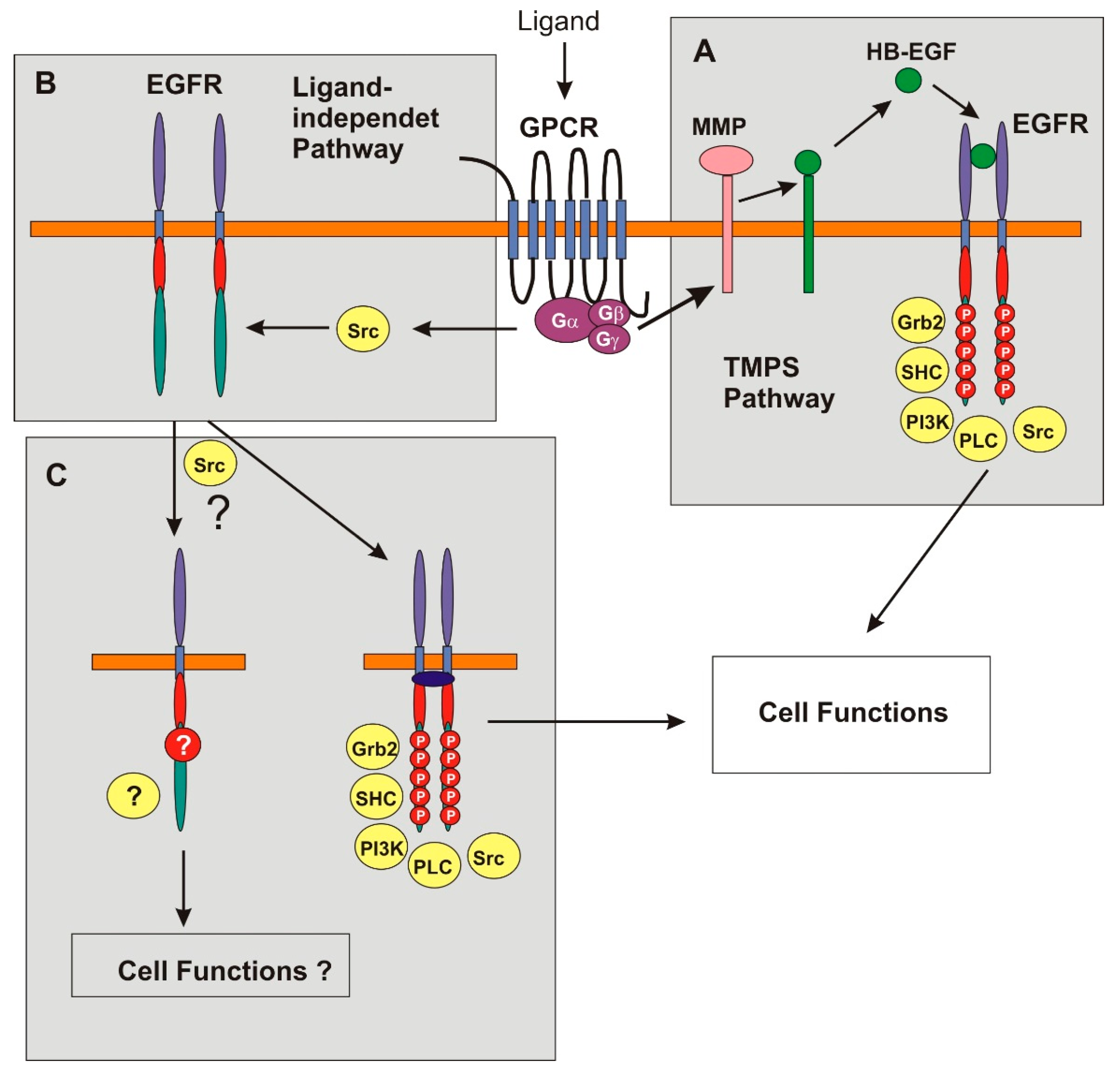
4. Transactivation of EGFR by GPCRs

5. Progress in the Last Five Years
6. Challenges and Future Research
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Heitzler, D.; Crepieux, P.; Poupon, A.; Clement, F.; Fages, F.; Reiter, E. Towards a systems biology approach of G protein-coupled receptor signalling: Challenges and expectations. Comptes Rendus Biol. 2009, 332, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Booe, J.M.; Pioszak, A.A. Structural insights into ligand recognition and selectivity for classes A, B, and C GPCRs. Eur. J. Pharmacol. 2015, 763, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. J. Virtual Libr. 2008, 13, 1857–1865. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; de Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by G protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal growth factor receptor transactivation: Mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharmacol. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hannan, R.D.; Thomas, W.G. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013, 280, 5258–5268. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Rostam, M.A.; Bernard, R.; Piva, T.J.; Mantri, N.; Guidone, D.; Zheng, W.; Osman, N.; Little, P.J. The expansion of GPCR transactivation-dependent signalling to include serine/threonine kinase receptors represents a new cell signalling frontier. Cell. Mol. Life Sci. 2015, 72, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Kolakowski, L.F., Jr. Gcrdb: A G-protein-coupled receptor database. Recept. Channels 1994, 2, 1–7. [Google Scholar] [PubMed]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Hoare, S.R. Mechanisms of peptide and nonpeptide ligand binding to class B G-protein-coupled receptors. Drug Discov. Today 2005, 10, 417–427. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu. Rev. Biochem. 1987, 56, 881–914. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; Goh, L.K. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 2009, 315, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Protein modules and signalling networks. Nature 1995, 373, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Protein-tyrosine kinases-new impressions of SRC and HCK. Nature 1997, 385, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta-Mol. Cell Res. 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wee, P.; Jiang, J.; Chen, X.; Wang, Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS ONE 2012, 7, e41354. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Kassis, J.; Solava, J.; Turner, T.; Lauffenburger, D.A. Growth factor-induced cell motility in tumor invasion. Acta Oncol. 2002, 41, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gluck, S.; Zhang, L.; Moran, M.F. Requirement for phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol. Cell. Biol. 1998, 18, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Wang, Z. Akt binds to and phosphorylates phospholipase C-γ1 in response to epidermal growth factor. Mol. Biol. Cell. 2006, 17, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Q.; Wang, Y.; Chen, X.; Wang, Z. PLC-γ1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 2009, 23, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the egf receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Wallasch, C.; Lankenau, A.; Herrlich, A.; Ullrich, A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997, 16, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef]
- Heeneman, S.; Haendeler, J.; Saito, Y.; Ishida, M.; Berk, B.C. Angiotensin II induces transactivation of two different populations of the platelet-derived growth factor β receptor. Key role for the p66 adaptor protein Shc. J. Biol. Chem. 2000, 275, 15926–15932. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Chao, M.V. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA 2001, 98, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Rajagopal, R.; Chao, M.V. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 2002, 13, 11–17. [Google Scholar] [CrossRef]
- Tu, H.; Xu, C.; Zhang, W.; Liu, Q.; Rondard, P.; Pin, J.P.; Liu, J. Gabab receptor activation protects neurons from apoptosis via IGF-1 receptor transactivation. J. Neurosci. 2010, 30, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Oligny-Longpre, G.; Corbani, M.; Zhou, J.; Hogue, M.; Guillon, G.; Bouvier, M. Engagement of beta-arrestin by transactivated insulin-like growth factor receptor is needed for V2 vasopressin receptor-stimulated ERK1/2 activation. Proc. Natl. Acad. Sci. USA 2012, 109, E1028–E1037. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.; Jin, Z.G.; Berk, B.C. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS). J. Biol. Chem. 2002, 277, 42997–43001. [Google Scholar] [CrossRef] [PubMed]
- Seye, C.I.; Yu, N.; Gonzalez, F.A.; Erb, L.; Weisman, G.A. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1). J. Biol. Chem. 2004, 279, 35679–35686. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.H.; Millette, E.; Kenagy, R.D.; Daum, G.; Clowes, A.W. Thrombin- and factor Xa-induced DNA synthesis is mediated by transactivation of fibroblast growth factor receptor-1 in human vascular smooth muscle cells. Circ. Res. 2004, 94, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. EGF receptor activation by GPCRs: An universal pathway reveals different versions. Mol. Cell. Endocrinol. 2011, 331, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 2001, 8, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Dempsey, P.J.; Eguchi, S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol. 2006, 291, C1–C10. [Google Scholar] [CrossRef] [PubMed]
- Etkovitz, N.; Tirosh, Y.; Chazan, R.; Jaldety, Y.; Daniel, L.; Rubinstein, S.; Breitbart, H. Bovine sperm acrosome reaction induced by G-protein-coupled receptor agonists is mediated by epidermal growth factor receptor transactivation. Dev. Biol. 2009, 334, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. Nadph-oxidase-dependent reactive oxygen species mediate egfr transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.H.; Hsiung, T.C.; Kuo, H.P.; Huang, C.D. Cross-talk between bradykinin and epidermal growth factor in regulating IL-6 production in human airway smooth muscle cells. Chang Gung Med. J. 2010, 33, 92–99. [Google Scholar] [PubMed]
- Cheng, C.Y.; Tseng, H.C.; Yang, C.M. Bradykinin-mediated cell proliferation depends on transactivation of EGF receptor in corneal fibroblasts. J. Cell. Physiol. 2012, 227, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Sancho, V.; di Florio, A.; Nuche-Berenguer, B.; Mantey, S.; Jensen, R.T. Bombesin receptor subtype-3 agonists stimulate the growth of lung cancer cells and increase EGF receptor tyrosine phosphorylation. Peptides 2011, 32, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Cho-Clark, M.; Larco, D.O.; Semsarzadeh, N.N.; Vasta, F.; Mani, S.K.; Wu, T.J. GnRH-(1–5) transactivates egfr in ishikawa human endometrial cells via an orphan G protein-coupled receptor. Mol. Endocrinol. 2014, 28, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Grundker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Terai, Y.; Kawaguchi, H.; Takai, M.; Yoo, S.; Tanaka, Y.; Tanaka, T.; Tsunetoh, S.; Sasaki, H.; Kanemura, M.; et al. GPR30 regulates the EGFR-Akt cascade and predicts lower survival in patients with ovarian cancer. J. Ovarian Res. 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Kurt, A.H.; Tiftik, R.N.; Un, I.; Ulker, S.; Buyukafsar, K. G protein-coupled estrogen receptor1 (GPER1) may mediate Rho-kinase (rock-2) up-regulation in coronary endothelial cells. Endocr. Regul. 2013, 47, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.J.; Seok, Y.M.; Arterburn, J.B.; Olatunji, L.A.; Kim, I.K. GPER-1 agonist G1 induces vasorelaxation through activation of epidermal growth factor receptor-dependent signalling pathway. J. Pharm. Pharmacol. 2013, 65, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Deng, X.; Wu, C.; Zhou, Q.; Chen, L.; Shi, Y.; Huang, H.; Zhou, N. Distinct kinetic and spatial patterns of protein kinase C (PKC)- and epidermal growth factor receptor (EGFR)-dependent activation of extracellular signal-regulated kinases 1 and 2 by human nicotinic acid receptor GPR109A. J. Biol. Chem. 2011, 286, 31199–31212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, C.; Li, H.; Li, C.; Hou, Q.; Liu, M.; Dong Xda, E.; Tu, L. GPR48-induced keratinocyte proliferation occurs through HB-EGF mediated EGFR transactivation. FEBS Lett. 2010, 584, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Cui, H.; Liu, S.; Qian, Y.; Wu, H.; Li, L.; Guan, Y.; Guan, X.; Zhang, L.; Fan, H.Y.; et al. LGR4 gene regulates corpus luteum maturation through modulation of the WNT-mediated EGFR-ERK signaling pathway. Endocrinology 2014, 155, 3624–3637. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, S.; Furuta, D.; Sugita, K.; Taniura, H.; Fujita, N. GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur. J. Pharmacol. 2013, 715, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Perrino, C.; Cannavo, A.; Schiattarella, G.G.; Borgia, F.; Sannino, A.; Pironti, G.; Gargiulo, G.; di Serafino, L.; Franzone, A.; et al. EGFR trans-activation by urotensin II receptor is mediated by β-arrestin recruitment and confers cardioprotection in pressure overload-induced cardiac hypertrophy. Basic Res. Cardiol. 2011, 106, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moyano, M.; Diaz, I.; Dionisio, N.; Zhang, X.; Avila-Medina, J.; Calderon-Sanchez, E.; Trebak, M.; Rosado, J.A.; Ordonez, A.; Smani, T. Urotensin-II promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation. Cardiovasc. Res. 2013, 100, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ulu, N.; Henning, R.H.; Guner, S.; Zoto, T.; Duman-Dalkilic, B.; Duin, M.; Gurdal, H. Intracellular transactivation of epidermal growth factor receptor by α1A-adrenoceptor is mediated by phosphatidylinositol 3-kinase independently of activation of extracellular signal regulated kinases 1/2 and serine-threonine kinases in chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 2013, 347, 47–56. [Google Scholar] [PubMed]
- Kuzumaki, N.; Suzuki, A.; Narita, M.; Hosoya, T.; Nagasawa, A.; Imai, S.; Yamamizu, K.; Morita, H.; Suzuki, T.; Okada, Y.; et al. Multiple analyses of G-protein coupled receptor (GPCR) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS ONE 2012, 7, e44368. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhong, S.; Ye, X.; Liao, Y.; Yao, F.; Yang, X.; Sun, B.; Zhang, J.; Li, Q.; Gao, Y.; et al. EGFR phosphorylates and inhibits lung tumor suppressor GPRC5A in lung cancer. Mol. Cancer 2014, 13, 233. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Mantey, S.A.; Moreno, P.; Nakamura, T.; Lacivita, E.; Leopoldo, M.; Jensen, R.T. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides 2015, 64, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.; Law, J.; Cvetkovic, D.D.; Pampillo, M.; McColl, L.; Pape, C.; di Guglielmo, G.M.; Postovit, L.M.; Babwah, A.V.; Bhattacharya, M. GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS ONE 2011, 6, e21599. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wadham, C.; Xia, P. Estrogen defines the dynamics and destination of transactivated EGF receptor in breast cancer cells: Role of S1P(3) receptor and Cdc42. Exp. Cell Res. 2013, 319, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Brusevold, I.J.; Tveteraas, I.H.; Aasrum, M.; Odegard, J.; Sandnes, D.L.; Christoffersen, T. Role of LPAR3, PKC and EGFR in LPA-induced cell migration in oral squamous carcinoma cells. BMC Cancer 2014, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Fujino, H.; Otake, S.; Seira, N.; Regan, J.W.; Murayama, T. Induction of cyclooxygenase-2 expression by prostaglandin E2 stimulation of the prostanoid EP4 receptor via coupling to galphai and transactivation of the epidermal growth factor receptor in HCA-7 human colon cancer cells. Eur. J. Pharmacol. 2013, 718, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Tveteraas, I.H.; Aasrum, M.; Brusevold, I.J.; Odegard, J.; Christoffersen, T.; Sandnes, D. Lysophosphatidic acid induces both EGFR-dependent and EGFR-independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, S.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. Thrombin induces epidermal growth factor receptor transactivation and CCL2 expression in human osteoblasts. Arthritis Rheum. 2012, 64, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Zhu, Z.; Jiang, W.; Xu, C. Transactivation of epidermal growth factor receptor through platelet-activating factor/receptor in ovarian cancer cells. J. Exp. Clin. Cancer Res. CR 2014, 33, 856. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, X.; Hong, S.; Zhang, M.; Cai, Q.; Jiang, W.; Xu, C. Epidermal growth factor induces platelet-activating factor production through receptors transactivation and cytosolic phospholipase A2 in ovarian cancer cells. J. Ovarian Res. 2014, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Tveteraas, I.H.; Muller, K.M.; Aasrum, M.; Odegard, J.; Dajani, O.; Guren, T.; Sandnes, D.; Christoffersen, T. Mechanisms involved in PGE2-induced transactivation of the epidermal growth factor receptor in MH1C1 hepatocarcinoma cells. J. Exp. Clin. Cancer Res. 2012, 31, 72. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Thomas, S.M.; Freilino, M.; Joyce, S.; Sahu, A.; Maxwell, J.; Argiris, A.; Seethala, R.; Grandis, J.R. Targeting GPCR-mediated P70S6K activity may improve head and neck cancer response to cetuximab. Clin. Cancer Res. 2011, 17, 4996–5004. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, S.; Kozono, D.; Ng, K.; Futalan, D.; Shen, Y.; Akers, J.C.; Steed, T.; Kushwaha, D.; Schlabach, M.; et al. Genome-wide shrna screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 2014, 5, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Ramachandran, R.; Hollenberg, M.D.; Muruve, D.A. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-β receptor signaling pathways contributes to renal fibrosis. J. Biol. Chem. 2013, 288, 37319–37331. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Yousif, M.H.; Dhaunsi, G.S.; Chandrasekhar, B.; Al-Farsi, O.; Benter, I.F. Angiotensin-(1–7) inhibits epidermal growth factor receptor transactivation via a mas receptor-dependent pathway. Br. J. Pharmacol. 2012, 165, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Chandrasekhar, B.; Attur, S.; Dhaunsi, G.S.; Yousif, M.H.; Benter, I.F. Transactivation of ErbB family of receptor tyrosine kinases is inhibited by angiotensin-(1–7) via its mas receptor. PLoS ONE 2015, 10, e0141657. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Yin, H.; Liao, Y.; Yao, F.; Li, Q.; Zhang, J.; Jiao, H.; Zhao, Y.; Xu, D.; Liu, S.; et al. Lung tumor suppressor GPRC5A binds EGFR and restrains its effector signaling. Cancer Res. 2015, 75, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Watt, H.L.; Kharmate, G.D.; Kumar, U. Somatostatin receptors 1 and 5 heterodimerize with epidermal growth factor receptor: Agonist-dependent modulation of the downstream MAPK signalling pathway in breast cancer cells. Cell Signal. 2009, 21, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.D.; Godfrey, C.B.; Niklas, C.; Canals, M.; Kocan, M.; Poole, D.P.; Murphy, J.E.; Alemi, F.; Cottrell, G.S.; Korbmacher, C.; et al. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J. Biol. Chem. 2013, 288, 22942–22960. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, A.M.; Baker, O.J.; Camden, J.M.; Rikka, S.; Petris, M.J.; Seye, C.I.; Erb, L.; Weisman, G.A. P2Y2 nucleotide receptors mediate metalloprotease-dependent phosphorylation of epidermal growth factor receptor and ErbB3 in human salivary gland cells. J. Biol. Chem. 2010, 285, 7545–7555. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Jiang, M.; Chen, L.; Xu, C.; Zhang, W.; Zhao, H.; Sun, B.; Xu, X.; Nan, F.; et al. An activity-based probe reveals dynamic protein-protein interactions mediating IGF-1R transactivation by the GABA(B) receptor. Biochem. J. 2012, 443, 627–634. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Purdue, B.W.; Gould, C.M.; Thomas, D.W.; Handoko, Y.; Qian, H.; Quaife-Ryan, G.A.; Morgan, K.A.; Simpson, K.J.; Thomas, W.G.; et al. A functional sirna screen identifies genes modulating angiotensin II-mediated EGFR transactivation. J. Cell Sci. 2013, 126, 5377–5390. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.L.; Godin, A.G.; Dore, K.; Freland, L.; Bouchard, N.; Nimmo, C.; Sergeev, M.; de Koninck, Y.; Wiseman, P.W.; Beaulieu, J.M. Quantification of receptor tyrosine kinase transactivation through direct dimerization and surface density measurements in single cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7016–7021. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 2013, 14, 10761–10790. [Google Scholar] [CrossRef] [PubMed]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Xu, C.; Keum, Y.S.; Reddy, B.; Conney, A.; Kong, A.N. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with β-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006, 27, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Amorino, G.P.; Deeble, P.D.; Parsons, S.J. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 2007, 26, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.M.; Bhattacharya, S.; Johnson, L.R. EGFR plays a pivotal role in the regulation of polyamine-dependent apoptosis in intestinal epithelial cells. Cell Signal. 2007, 19, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. TGF-beta1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-Src)/EGFR(y845) and Rho/rock signaling. J. Mol. Cell. Cardiol. 2008, 44, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Wettstein, M.; Haussinger, D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology 2004, 127, 1540–1557. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Eberle, A.; Grether-Beck, S.; Haussinger, D. Involvement of nadph oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J. Biol. Chem. 2005, 280, 27179–27194. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wang, Y.T.; Tzeng, D.W.; Yang, J.L. Lead acetate induces EGFR activation upstream of SFK and PKCalpha linkage to the Ras/Raf-1/ERK signaling. Toxicol. Appl. Pharmacol. 2009, 235, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N.; Tojo, A.; Hino, M.; Yazaki, Y.; Shibuya, M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 1992, 186, 768–774. [Google Scholar] [CrossRef]
- Song, H.; Huang, L.; Zhang, M.; Wang, X.; Song, S.; Yang, L. Transphosphorylation of EGFR at Y845 plays an important role in its autophosphorylation and kinase activity. Oncol. Rep. 2014, 31, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 activates mitogen-activated protein kinase/ERK pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Bhola, N.E.; Zhang, Q.; Contrucci, S.C.; Wentzel, A.L.; Freilino, M.L.; Gooding, W.E.; Siegfried, J.M.; Chan, D.C.; Grandis, J.R. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 11831–11839. [Google Scholar] [CrossRef] [PubMed]
- Piiper, A.; Elez, R.; You, S.J.; Kronenberger, B.; Loitsch, S.; Roche, S.; Zeuzem, S. Cholecystokinin stimulates extracellular signal-regulated kinase through activation of the epidermal growth factor receptor, yes, and protein kinase C. Signal amplification at the level of Raf by activation of protein kinase cepsilon. J. Biol. Chem. 2003, 278, 7065–7072. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhou, Z.; Shan, L.; Zeng, H.; Hua, Y.; Cai, Z. The importance of Src signaling in sarcoma. Oncol. Lett. 2015, 10, 17–22. [Google Scholar] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research. Int. J. Mol. Sci. 2016, 17, 95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010095
Wang Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research. International Journal of Molecular Sciences. 2016; 17(1):95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010095
Chicago/Turabian StyleWang, Zhixiang. 2016. "Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research" International Journal of Molecular Sciences 17, no. 1: 95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010095