
Transcriptome-Wide Identification and Prediction of miRNAs and Their Targets in Paris polyphylla var. yunnanensis by High-Throughput Sequencing Analysis

 ,
,
Abstract
:1. Introduction
2. Results
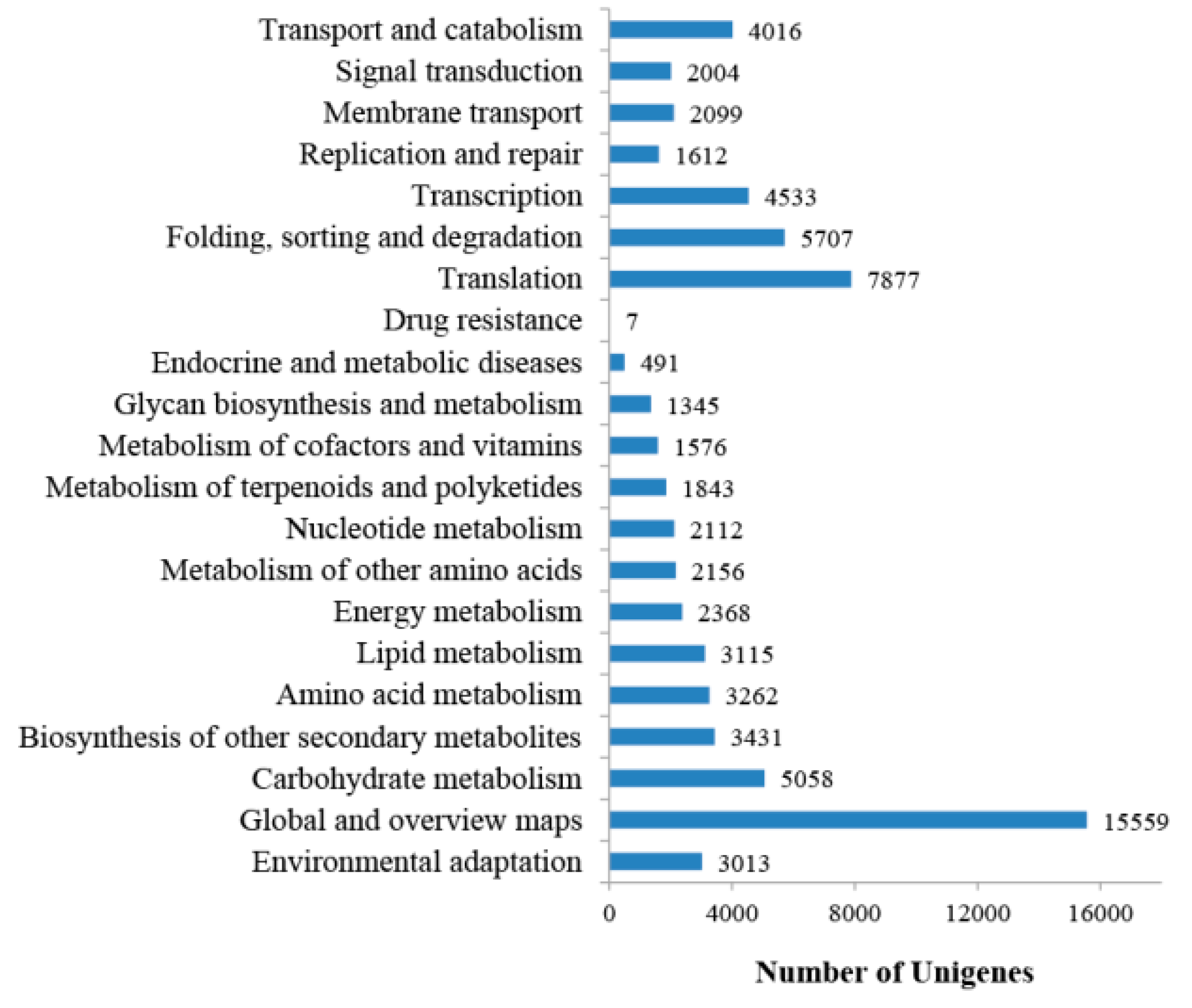
2.1. Transcriptome Sequencing, Assembly, and Annotation
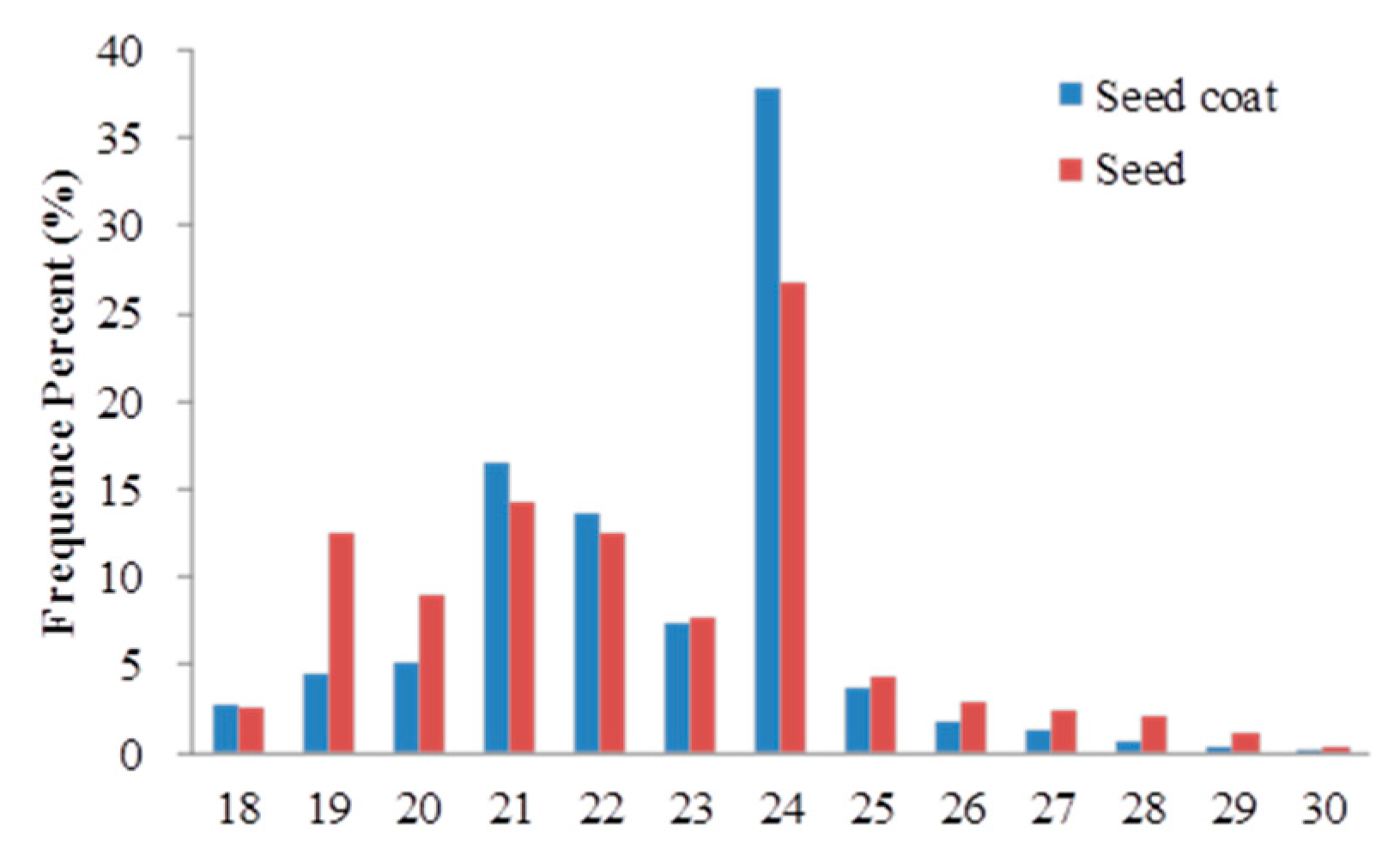
2.2. Overview of Small RNA Sequencing and Annotation
2.3. Conserved miRNAs in P. polyphylla var. yunnanensis
2.4. Novel miRNAs in P. polyphylla var. yunnanensis
2.5. Target Prediction of Novel and Conserved miRNAs
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Preparation, Library Construction, and Illumina Sequencing
4.3. Transcript Assembly and Annotation
4.4. Bioinformatics Analysis of sRNAs
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Ann. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Lindow, M.; Gorodkin, J. Principles and limitations of computational microRNA gene and target finding. DNA Cell Biol. 2007, 26, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Park, S.; Zhai, J.; Gurazada, S.G.; de Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Wei, S.; Bradford, K.J. Delay of germination1 (DOG1) regulates both seed dormancy and flowering time through microRNA pathways. Proc. Natl. Acad. Sci. USA 2016, 113, E2199–E2206. [Google Scholar] [CrossRef] [PubMed]
- Li, H. The phylogeny of the genus Paris, L. Acta Bot. Yunnanica 1984, 6, 351–362. [Google Scholar]
- Li, Y. Studies on the introduction cultivation of genus Paris L. I. A premilinary report on sexual propogation of Paris polyphylla var. yunnanensis. Acta Bot. Yunnanica 1982, 4, 429–431. [Google Scholar]
- Chen, W.; Yang, Y.; Ma, S.; Li, J. Study on Dormancy Type of P. polyphylla var. yunnanensis Seeds. Southwest China J. Agric. Sci. 2015, 28, 783–786. [Google Scholar]
- Huang, W.; Meng, F.; Zhang, W.; Wang, Y. Study on seed dormancy mechanism of P. polyphylla var. yunnanensis. Chin. Agric. Sci. Bull. 2008, 24, 242–246. [Google Scholar]
- Zhou, L.; Wu, J.; Wang, S. Low-temperature stratification strategies and growth regulators for rapid induction of Paris polyphylla var. yunnanensis seed germination. Plant Growth Regul. 2003, 41, 179–183. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, C.; Yang, L.; Lu, L.; He, X. Effects of temperature and gibberellin treatments on the second growth of seeds of Paris polyphylla var. yunnanensis. Seed 2003, 33–34. [Google Scholar]
- Yu, X.D.; Zhang, P.; Song, F.J.; Liu, J.; Xi, X.Y.; Yu, P.F. Effect of Paris seed coat extract on seed germination and seedling growth of rice. Jiangsu Agric. Sci. 2016, 44, 96–98. [Google Scholar]
- Royal Botanic Gardens, Kew. Available online: www.kew.org/cval/homepage.html (accessed on 19 January 2017).
- Zonneveld, B.J.; Leitch, I.J.; Bennett, M.D. First nuclear DNA amounts in more than 300 angiosperms. Ann. Bot. 2005, 96, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.P.; Yu, K.; Zhao, Y.; Liu, Y.X.; Yu, H.S.; Pang, X.; Xiong, C.Q.; Tan, D.W.; Gao, Y.; Liu, C.; et al. Characterization of steroidal glycosides from the extract of Paris Polyphylla var. Yunnanensis by UPLC/Q-TOF MSE. J. Pharm. Biomed. Anal. 2012, 62, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Gao, W.; Zhang, Y.; Wang, Y. A new phenylpropanoid glycosides from Paris polyphylla var. yunnanensis. Fitoterapia 2008, 79, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, H.; Zhang, Z.; Hu, X.; Li, H. Medicinal values and their chemical bases of Paris. China J. Chin. Mater. Medica 2015, 40, 833–839. [Google Scholar]
- Qi, J.; Zheng, N.; Zhang, B.; Sun, P.; Hu, S.; Xu, W.; Ma, Q.; Zhao, T.; Zhou, L.; Qin, M.; et al. Mining genes involved in the stratification of Paris polyphylla seeds using high-throughput embryo transcriptome sequencing. BMC Genom. 2013, 14, 358. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gao, L.; Wang, H.; Chen, X.; Wang, Y.; Yang, H.; Wei, C.; Wan, X.; Xia, T. The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct. Integr. Genom. 2013, 13, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Liang, D.; Zhang, Z.; Yin, W.; Xia, X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genom. 2013, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Qiu, D.; Wilson, I.W.; Zhao, H.; Lu, S.; Miao, J.; Feng, S.; Bai, L.; Wu, Q.; Tu, D.; Ma, X.; Tang, Q. Identification of novel and conserved microRNAs in Panax notoginseng roots by high-throughput sequencing. BMC Genom. 2015, 16, 835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; Ling, L.Z.; Yi, T.S. Evolution and divergence of SBP-box genes in land plants. BMC Genom. 2015, 16, 787. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Dahal, P.; Kunusoth, K.; McCallum, C.M.; Bradford, K.J. Expression of 9-cis-epoxycarotenoid dioxygenase4 is essential for thermoinhibition of lettuce seed germination but not for seed development or stress tolerance. Plant Cell 2013, 25, 884–900. [Google Scholar] [CrossRef] [PubMed]
- Argyris, J.; Dahal, P.; Hayashi, E.; Still, D.W.; Bradford, K.J. Genetic variation for lettuce seed thermoinhibition is associated with temperature-sensitive expression of abscisic Acid, gibberellin, and ethylene biosynthesis, metabolism, and response genes. Plant Physiol. 2008, 148, 926–947. [Google Scholar] [CrossRef] [PubMed]
- Omidbakhshfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-regulating factors (GRFs): A small transcription factor family with important functions in plant biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Zhang, S.D.; Ling, L.Z. De novo transcriptome analysis to identify flavonoid biosynthesis genes in Stellera chamaejasme. Plant Gene 2015, 4, 64–68. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nature Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends. Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sequences | Samples | |
|---|---|---|
| Seed Coat | Seed | |
| Reads | ||
| Total Raw Reads (MB) | 63.33 | 63.33 |
| Total Clean Reads (MB) | 59.42 | 59.44 |
| Total Clean Reads (MB) | 59.42 | 59.44 |
| Total Clean Bases (GB) | 8.91 | 8.92 |
| Clean Reads Q 20 (%) | 97.21 | 97.42 |
| Unigenes | ||
| Number | 135,008 | 90,419 |
| Mean Length | 955 | 1241 |
| Total unigenes | 14,6671 | |
| Reads | Samples | ||
|---|---|---|---|
| Seed Coat | Seed | Total | |
| Raw reads | 13,045,488 | 12,651,991 | |
| High quality | 13,006,850 | 12,606,706 | |
| 3′ Adaptor null | 45,875 | 42,137 | |
| Insert null | 2433 | 608 | |
| 5′ Adaptor contaminants | 13,837 | 6134 | |
| Smaller than 18 nt | 500,459 | 126,650 | |
| Clean reads | 12,444,074 | 12,431,325 | |
| Conserved miRNAs | 219 | 148 | 263 |
| Novel miRNAs | 902 | 640 | 768 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, L.-Z.; Zhang, S.-D.; Zhao, F.; Yang, J.-L.; Song, W.-H.; Guan, S.-M.; Li, X.-S.; Huang, Z.-J.; Cheng, L. Transcriptome-Wide Identification and Prediction of miRNAs and Their Targets in Paris polyphylla var. yunnanensis by High-Throughput Sequencing Analysis. Int. J. Mol. Sci. 2017, 18, 219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010219
Ling L-Z, Zhang S-D, Zhao F, Yang J-L, Song W-H, Guan S-M, Li X-S, Huang Z-J, Cheng L. Transcriptome-Wide Identification and Prediction of miRNAs and Their Targets in Paris polyphylla var. yunnanensis by High-Throughput Sequencing Analysis. International Journal of Molecular Sciences. 2017; 18(1):219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010219
Chicago/Turabian StyleLing, Li-Zhen, Shu-Dong Zhang, Fan Zhao, Jin-Long Yang, Wen-Hui Song, Shen-Min Guan, Xin-Shu Li, Zhuang-Jia Huang, and Le Cheng. 2017. "Transcriptome-Wide Identification and Prediction of miRNAs and Their Targets in Paris polyphylla var. yunnanensis by High-Throughput Sequencing Analysis" International Journal of Molecular Sciences 18, no. 1: 219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010219