Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models

 , and
, and
Abstract
:1. Introduction
2. Glioma Cell Invasion: The Picture of a Complex Event
2.1. How and Where Glioma Cells Move
2.2. The Actors Involved in Glioma Cell Invasion
2.2.1. Tumour Cells
2.2.2. The Tumour Microenvironment
The Extracellular Matrix
Interstitial Flow
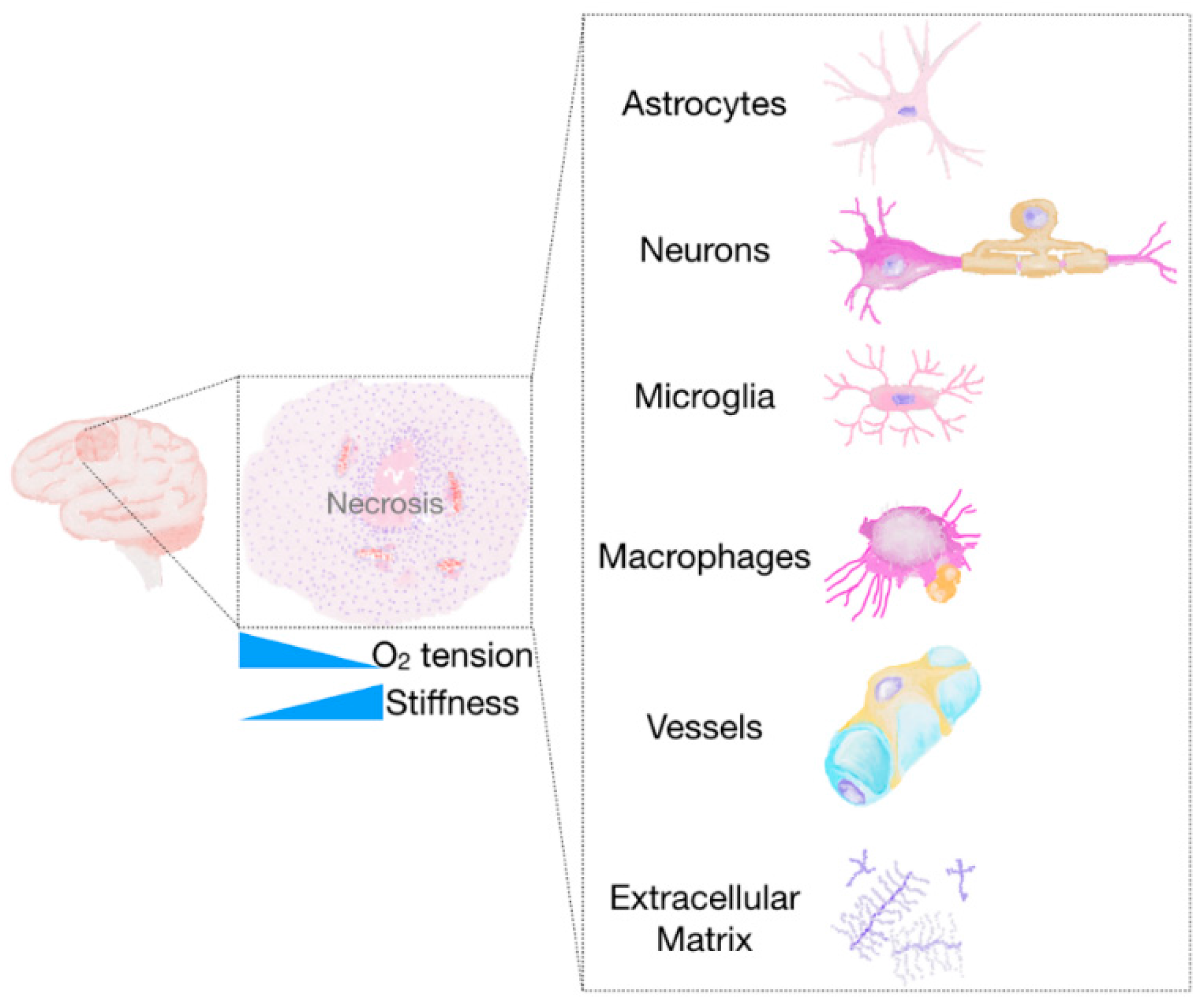
Hypoxia and Acidosis
Non-Neoplastic Cells of the Glioma Microenvironment
3. Evolution of In Vitro Models to Study Glioma Invasion
3.1. From 2D Models to 3D Models
3.1.1. Monolayer Culture and Scratch Assays
3.1.2. Transwell Assays
3.2. 3D Models: Taking into Account Cell-ECM and Cell-Cell Interactions
3.2.1. Ex Vivo Tumour Sections
3.2.2. 3D Invasion Model
3.2.3. Modified Transwell Assays
3.2.4. Spheroids
3.3. Engineered Models
3.3.1. Stiffness, Confinement, ECM and Chemotactic Gradients in 2D
3.3.2. Engineered 3D Models
Stiffness
ECM Composition
3.3.3. Migration along Constrained Paths
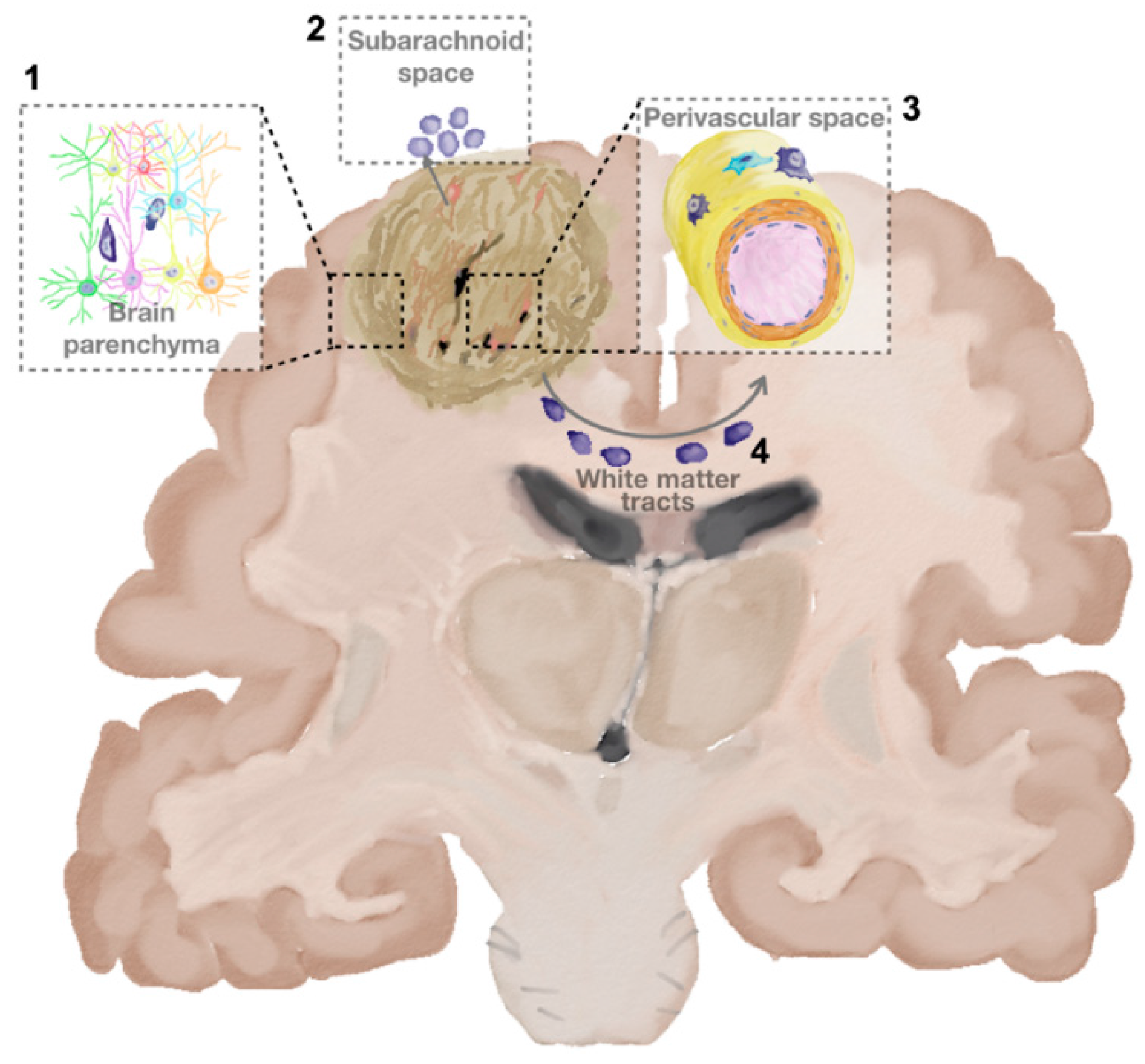
Parenchyma Invasion
Perivascular Invasion
3.3.4. Interstitial Flow
3.3.5. Cell-Cell Interaction
3.4. Towards Patient-Based Assays
4. Summary and Future Perspectives
5. Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C.K. Recurrent glioblastoma multiforme: A review of natural history and management options. Neurosurg. Focus 2006, 20, E5. [Google Scholar] [CrossRef] [PubMed]
- Lafitte, F.; Morel-Precetti, S.; Martin-Duverneuil, N.; Guermazi, A.; Brunet, E.; Heran, F.; Chiras, J. Multiple glioblastomas: Ct and MR features. Eur. Radiol. 2001, 11, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Capper, D.; Jeibmann, A.; Habel, A.; Paulus, W.; Troost, D.; von Deimling, A. Addressing diffuse glioma as a systemic brain disease with single-cell analysis. Arch. Neurol. 2012, 69, 523–526. [Google Scholar] [PubMed]
- Soffietti, R.; Baumert, B.G.; Bello, L.; von Deimling, A.; Duffau, H.; Frenay, M.; Grisold, W.; Grant, R.; Graus, F.; Hoang-Xuan, K.; et al. Guidelines on management of low-grade gliomas: Report of an efns-eano task force. Eur. J. Neurol. 2010, 17, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Ius, T.; Isola, M.; Budai, R.; Pauletto, G.; Tomasino, B.; Fadiga, L.; Skrap, M. Low-grade glioma surgery in eloquent areas: Volumetric analysis of extent of resection and its impact on overall survival. A single-institution experience in 190 patients: Clinical article. J. Neurosurg. 2012, 117, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Bolteus, A.J.; Berens, M.E.; Pilkington, G.J. Migration and invasion in brain neoplasms. Curr. Neurol. Neurosci. Rep. 2001, 1, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro tumor models: Advantages, disadvantages, variables and selecting the right platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Sohrabi, A.; Seidlits, S.K. Integrating the glioblastoma microenvironment into engineered experimental models. Future Sci. OA 2017, 3, FSO189. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P. Extra-neural metastases of malignant gliomas: Myth or reality? Cancers 2011, 3, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Beadle, C.; Assanah, M.C.; Monzo, P.; Vallee, R.; Rosenfeld, S.S.; Canoll, P. The role of myosin II in glioma invasion of the brain. Mol. Biol. Cell 2008, 19, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E. Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 2005, 15, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Cayre, M.; Canoll, P.; Goldman, J.E. Cell migration in the normal and pathological postnatal mammalian brain. Prog. Neurobiol. 2009, 88, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.J. Structural development in gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Tam, E.M.; Wu, Y.I.; Butler, G.S.; Stack, M.S.; Overall, C.M. Collagen binding properties of the membrane type1 matrix metalloproteinase (MT1-MMP) hemopexin C domain. The ectodomain of the 44-kDa autocatalytic product of MT1-MMP inhibits cell invasion by disrupting native type I collagen cleavage. J. Biol. Chem. 2002, 277, 39005–39014. [Google Scholar] [CrossRef] [PubMed]
- Wear, M.A.; Schafer, D.A.; Cooper, J.A. Actin dynamics: Assembly and disassembly of actin networks. Curr. Biol. 2000, 10, R891–R895. [Google Scholar] [CrossRef]
- Roos, A.; Ding, Z.; Loftus, J.C.; Tran, N.L. Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Front. Oncol. 2017, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Persano, L.; Rampazzo, E.; Basso, G.; Viola, G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Hsieh, I.-Y.; Huang, X.; Li, J.; Zhao, W. Glioblastoma stem-like cells: Characteristics, microenvironment and therapy. Front. Pharmacol. 2016, 7, 477. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Swartz, M.A. Modeling tumor microenvironments in vitro. J. Biomech. Eng. 2014, 136, 021011. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qin, Z.; Chen, Z.; Xie, L.; Wang, R.; Zhao, H. Tumor microenvironment in treatment of glioma. Open Med. 2017, 12, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brösicke, N.; van Landeghem, F.K.H.; Scheffler, B.; Faissner, A. Tenascin-c is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013, 354, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Kwak, H.J.; Lee, S.H. Role of hyaluronan in glioma invasion. Cell. Adhes. Migr. 2008, 2, 202–207. [Google Scholar] [CrossRef]
- Kim, Y.; Kumar, S. Cd44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, J.; Wu, H.; Sang, Y.; Shi, X.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Eberhart, C.G.; Laterra, J.; Ying, M. Hmmr maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2014, 74, 3168–3179. [Google Scholar] [CrossRef] [PubMed]
- Delpech, B.; Maingonnat, C.; Girard, N.; Chauzy, C.; Maunoury, R.; Olivier, A.; Tayot, J.; Creissard, P. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur. J. Cancer 1993, 29A, 1012–1017. [Google Scholar] [CrossRef]
- Brösicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Joester, A.; Faissner, A. The structure and function of tenascins in the nervous system. Matrix Biol. 2001, 20, 13–22. [Google Scholar] [CrossRef]
- Solis, M.A.; Chen, Y.H.; Wong, T.Y.; Bittencourt, V.Z.; Lin, Y.C.; Huang, L.L. Hyaluronan regulates cell behavior: A potential niche matrix for stem cells. Biochem. Res. Int. 2012, 2012, 346972. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, P.; Yang, L.; Yu, X.; Ye, X.; Yang, J.; Qian, C.; Zhang, X.; Cui, Y.H.; Bian, X.W. Activation of toll-like receptor 2 promotes invasion by upregulating mmps in glioma stem cells. Am. J. Transl. Res. 2015, 7, 607–615. [Google Scholar] [PubMed]
- Inoue, A.; Takahashi, H.; Harada, H.; Kohno, S.; Ohue, S.; Kobayashi, K.; Yano, H.; Tanaka, J.; Ohnishi, T. Cancer stem-like cells of glioblastoma characteristically express MMP-13 and display highly invasive activity. Int. J. Oncol. 2010, 37, 1121–1131. [Google Scholar] [PubMed]
- Chen, X.; Chen, L.; Chen, J.; Hu, W.; Gao, H.; Xie, B.; Wang, X.; Yin, Z.; Li, S.; Wang, X. Adam17 promotes u87 glioblastoma stem cell migration and invasion. Brain Res. 2013, 1538, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. Adam-9 is a novel mediator of tenascin-c-stimulated invasiveness of brain tumor-initiating cells. Neuro Oncol. 2015, 17, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Courtneidge, S.A. The adams family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gole, B.; Huszthy, P.C.; Popovic, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Weaver, V.M. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.J.; Valdeolmillos, M. Actomyosin contraction at the cell rear drives nuclear translocation in migrating cortical interneurons. J. Neurosci. 2010, 30, 8660–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannone, G.; Rondé, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3d reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Sontheimer, H. Hydrodynamic cellular volume changes enable glioma cell invasion. J. Neurosci. 2011, 31, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Bellamkonda, R.V.; Swartz, M.A. Interstitial flow in a 3d microenvironment increases glioma invasion by a cxcr4-dependent mechanism. Cancer Res. 2013, 73, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, K.M.; Logsdon, D.K.; Floyd, D.H.; Peirce, S.M.; Purow, B.W.; Munson, J.M. Interstitial flow differentially increases patient-derived glioblastoma stem cell invasion via cxcr4, cxcl12 and cd44-mediated mechanisms. Integr. Biol. 2016, 8, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Hara, A.; Yasui, Y.; Kumabe, T.; Matsumoto, T.; Saegusa, M. Hypoxia-mediated cancer stem cells in pseudopalisades with activation of hypoxia-inducible factor-1alpha/akt axis in glioblastoma. Hum. Pathol. 2015, 46, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Talasila, K.M.; Rosland, G.V.; Hagland, H.R.; Eskilsson, E.; Flones, I.H.; Fritah, S.; Azuaje, F.; Atai, N.; Harter, P.N.; Mittelbronn, M.; et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. 2017, 19, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Mendez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of hif-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the cd133-positive glioma stem cells through activation of hif-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. Hif-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Seidel, S.; Bogurcu, N.; Graf, S.; Garvalov, B.K.; Acker, T. Acidosis acts through hsp90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res. 2016, 76, 5845–5856. [Google Scholar] [CrossRef] [PubMed]
- Cong, D.; Zhu, W.; Shi, Y.; Pointer, K.B.; Clark, P.A.; Shen, H.; Kuo, J.S.; Hu, S.; Sun, D. Upregulation of nhe1 protein expression enables glioblastoma cells to escape tmz-mediated toxicity via increased h(+) extrusion, cell migration and survival. Carcinogenesis 2014, 35, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, I.; Thanos, S.; Paulus, W. Microglia promote glioma migration. Acta Neuropathol. 2002, 103, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef] [PubMed]
- A Dzaye, O.D.; Hu, F.; Derkow, K.; Haage, V.; Euskirchen, P.; Harms, C.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma stem cells but not bulk glioma cells upregulate IL-6 secretion in microglia/brain macrophages via toll-like receptor 4 signaling. J. Neuropathol. Exp. Neurol. 2016, 75, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; van Rooijen, N.; Kiwit, J.; Synowitz, M.; Glass, R.; Kettenmann, H. Minocycline reduces glioma expansion and invasion by attenuating microglial mt1-mmp expression. Brain Behav. Immun. 2011, 25, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial mt1-mmp expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumour Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Tran, N.L.; Menashi, E.; Rohl, C.; Kloss, J.; Bay, R.C.; Berens, M.E.; Loftus, J.C. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia 2005, 7, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Loftus, J.C.; Yang, Z.; Kloss, J.; Dhruv, H.; Tran, N.L.; Riggs, D.L. A novel interaction between pyk2 and map4k4 is integrated with glioma cell migration. J. Signal Transduct. 2013, 2013, 956580. [Google Scholar] [CrossRef] [PubMed]
- Rolon-Reyes, K.; Kucheryavykh, Y.V.; Cubano, L.A.; Inyushin, M.; Skatchkov, S.N.; Eaton, M.J.; Harrison, J.K.; Kucheryavykh, L.Y. Microglia activate migration of glioma cells through a pyk2 intracellular pathway. PLoS ONE 2015, 10, e0131059. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekstrom, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-cd44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via tgf-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Kato, M.; Yamashita, H.; Nister, M.; Miyazono, K.; Heldin, C.H.; Funa, K. Enhanced expression of transforming growth factor-beta and its type-I and type-II receptors in human glioblastoma. Int. J. Cancer 1995, 62, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of cx3cr1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly teamwork: Neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011, 8, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Samaras, V.; Piperi, C.; Korkolopoulou, P.; Zisakis, A.; Levidou, G.; Themistocleous, M.S.; Boviatsis, E.I.; Sakas, D.E.; Lea, R.W.; Kalofoutis, A.; et al. Application of the elispot method for comparative analysis of interleukin (il)-6 and il-10 secretion in peripheral blood of patients with astroglial tumors. Mol. Cell. Biochem. 2007, 304, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits m2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cossette, S.M.; Rarick, K.R.; Gershan, J.; Dwinell, M.B.; Harder, D.R.; Ramchandran, R. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS ONE 2013, 8, e80933. [Google Scholar] [CrossRef] [PubMed]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of astrocytes by glioma cells to facilitate invasiveness: A mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [PubMed]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015, 4, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Martin, V.; Fueyo, J.; Lee, O.H.; Xu, J.; Cortes-Santiago, N.; Alonso, M.M.; Aldape, K.; Colman, H.; Gomez-Manzano, C. Tie2/tek modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget 2010, 1, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Behnan, J.; Isakson, P.; Joel, M.; Cilio, C.; Langmoen, I.A.; Vik-Mo, E.O.; Badn, W. Recruited brain tumor-derived mesenchymal stem cells contribute to brain tumor progression. Stem Cells 2014, 32, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the il-6/gp130/stat3 pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Bourkoula, E.; Mangoni, D.; Ius, T.; Pucer, A.; Isola, M.; Musiello, D.; Marzinotto, S.; Toffoletto, B.; Sorrentino, M.; Palma, A.; et al. Glioma-associated stem cells: A novel class of tumor-supporting cells able to predict prognosis of human low-grade gliomas. Stem Cells 2014, 32, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Rape, A.; Ananthanarayanan, B.; Kumar, S. Engineering strategies to mimic the glioblastoma microenvironment. Adv. Drug Deliv. Rev. 2014, 79–80, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Belien, A.T.; Paganetti, P.A.; Schwab, M.E. Membrane-type 1 matrix metalloprotease (mt1-mmp) enables invasive migration of glioma cells in central nervous system white matter. J. Cell Biol. 1999, 144, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Loo, M.A.; Rief, M.D.; Tran, N.; Berens, M.E. Substrates for astrocytoma invasion. Neurosurgery 1995, 37, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Pilkington, G.J.; Merzak, A. Hyaluronic acid/cd44h interaction induces cell detachment and stimulates migration and invasion of human glioma cells in vitro. Int. J. Cancer 1995, 63, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Elkin, B.S.; Azeloglu, E.U.; Costa, K.D.; Morrison, B., 3rd. Mechanical heterogeneity of the rat hippocampus measured by atomic force microscope indentation. J. Neurotrauma 2007, 24, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Griffith, L.G.; Swartz, M.A. Capturing complex 3d tissue physiology in vitro. Nat. Rev. Mol. Cell Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling tissue morphogenesis and cancer in 3d. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschlager, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Able, R.A., Jr.; Dudu, V.; Vazquez, M. Migration and invasion of brain tumors. In Glioma—Exploring Its Biology and Practical Relevance; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, M.A.; Ulbricht, U.; Gruner, K.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Glioblastoma and cerebral microvascular endothelial cell migration in response to tumor-associated growth factors. Neurosurgery 2003, 52, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 2007, 99, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; Vande Woude, G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar] [PubMed]
- Moriyama, T.; Kataoka, H.; Seguchi, K.; Tsubouchi, H.; Koono, M. Effects of hepatocyte growth factor (HGF) on human glioma cells in vitro: HGF acts as a motility factor in glioma cells. Int. J. Cancer 1996, 66, 678–685. [Google Scholar] [CrossRef]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Lee, H.C.; Kim, M.S.; Lee, S.H.; Park, I.C.; Rhee, C.H.; Hong, S.I. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through src/epidermal growth factor receptor-mediated p38/akt and phosphatidylinositol 3-kinase/akt signaling pathways. Cancer Res. 2006, 66, 8511–8519. [Google Scholar] [CrossRef] [PubMed]
- Brekhman, V.; Neufeld, G. A novel asymmetric 3d in vitro assay for the study of tumor cell invasion. BMC Cancer 2009, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. Rhoa and rhoc have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.B.; Gamble, J.R.; Clark-Lewis, I.; Vadas, M.A. Interleukin-8 induces neutrophil transendothelial migration. Immunology 1991, 72, 65–72. [Google Scholar] [PubMed]
- Pignatelli, J.; Goswami, S.; Jones, J.G.; Rohan, T.E.; Pieri, E.; Chen, X.; Adler, E.; Cox, D.; Maleki, S.; Bresnick, A.; et al. Invasive breast carcinoma cells from patients exhibit menainv- and macrophage-dependent transendothelial migration. Sci. Signal. 2014, 7, ra112. [Google Scholar] [CrossRef] [PubMed]
- Rahn, J.J.; Chow, J.W.; Horne, G.J.; Mah, B.K.; Emerman, J.T.; Hoffman, P.; Hugh, J.C. Muc1 mediates transendothelial migration in vitro by ligating endothelial cell icam-1. Clin. Exp. Metastasis 2005, 22, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of kca3.1 and clc-3. J. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Schichor, C.; Kerkau, S.; Visted, T.; Martini, R.; Bjerkvig, R.; Tonn, J.C.; Goldbrunner, R. The brain slice chamber, a novel variation of the boyden chamber assay, allows time-dependent quantification of glioma invasion into mammalian brain in vitro. J. Neurooncol. 2005, 73, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Kim, H.W.; Lee, J.H.; Kang, S.S.; Rhu, H.H.; Jeong, Y.I.; Yang, S.Y.; Chung, H.Y.; Bae, C.S.; Choi, C.; et al. Brain tumor invasion model system using organotypic brain-slice culture as an alternative to in vivo model. J. Cancer Res. Clin. Oncol. 2002, 128, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Matsumura, H.; Izumoto, S.; Hiraga, S.; Hayakawa, T. A novel model of glioma cell invasion using organotypic brain slice culture. Cancer Res. 1998, 58, 2935–2940. [Google Scholar] [PubMed]
- Menyhárt, O.; Harami-Papp, H.; Sukumar, S.; Schäfer, R.; Magnani, L.; de Barrios, O.; Győrffy, B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1866, 300–319. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Kondo, J.; Endo, H.; Okuyama, H.; Ishikawa, O.; Iishi, H.; Tsujii, M.; Ohue, M.; Inoue, M. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6235–6240. [Google Scholar] [CrossRef] [PubMed]
- Bjerkvig, R.; Tonnesen, A.; Laerum, O.D.; Backlund, E.O. Multicellular tumor spheroids from human gliomas maintained in organ culture. J. Neurosurg. 1990, 72, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Nederman, T. A method to measure the radio and chemosensitivity of human spheroids. Adv. Exp. Med. Biol. 1983, 159, 399–417. [Google Scholar] [PubMed]
- Lund-Johansen, M.; Bjerkvig, R.; Humphrey, P.A.; Bigner, S.H.; Bigner, D.D.; Laerum, O.D. Effect of epidermal growth factor on glioma cell growth, migration and invasion in vitro. Cancer Res. 1990, 50, 6039–6044. [Google Scholar] [PubMed]
- Nederman, T.; Acker, H.; Carlsson, J. Penetration of substances into tumor tissue: A methodological study with microelectrodes and cellular spheroids. In Vitro 1983, 19, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Mahesparan, R.; Tysnes, B.B.; Read, T.A.; Enger, P.O.; Bjerkvig, R.; Lund-Johansen, M. Extracellular matrix-induced cell migration from glioblastoma biopsy specimens in vitro. Acta Neuropathol. 1999, 97, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.; Huettner, C.; Tonn, J.C. Collagens, integrins and the mesenchymal drift in glioblastomas: A comparison of biopsy specimens, spheroid and early monolayer cultures. Int. J. Cancer 1994, 58, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Engebraaten, O.; Bjerkvig, R.; Pedersen, P.H.; Laerum, O.D. Effects of EGF, bFGF, NGF and PDGF(bb) on cell proliferative, migratory and invasive capacities of human brain-tumour biopsies in vitro. Int. J. Cancer 1993, 53, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Van Tilborg, A.A.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Engebraaten, O.; Bjerkvig, R.; Lund-Johansen, M.; Wester, K.; Pedersen, P.H.; Mork, S.; Backlund, E.O.; Laerum, O.D. Interaction between human brain tumour biopsies and fetal rat brain tissue in vitro. Acta Neuropathol. 1990, 81, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Grundy, T.J.; De Leon, E.; Griffin, K.R.; Stringer, B.W.; Day, B.W.; Fabry, B.; Cooper-White, J.; O’Neill, G.M. Differential response of patient-derived primary glioblastoma cells to environmental stiffness. Sci. Rep. 2016, 6, 23353. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Kumar, S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc. Natl. Acad. Sci. USA 2012, 109, 10334–10339. [Google Scholar] [CrossRef] [PubMed]
- Umesh, V.; Rape, A.D.; Ulrich, T.A.; Kumar, S. Microenvironmental stiffness enhances glioma cell proliferation by stimulating epidermal growth factor receptor signaling. PLoS ONE 2014, 9, e101771. [Google Scholar] [CrossRef] [PubMed]
- Cretu, A.; Castagnino, P.; Assoian, R. Studying the effects of matrix stiffness on cellular function using acrylamide-based hydrogels. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.; Bacakova, L.; Newman, C.; Hategan, A.; Griffin, M.; Discher, D. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 2004, 86, 617–628. [Google Scholar] [CrossRef]
- Sen, S.; Dong, M.; Kumar, S. Isoform-specific contributions of alpha-actinin to glioma cell mechanobiology. PLoS ONE 2009, 4, e8427. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Ng, W.P.; Kumar, S. Contributions of talin-1 to glioma cell-matrix tensional homeostasis. J. R. Soc. Interface 2012, 9, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- MacKay, J.L.; Keung, A.J.; Kumar, S. A genetic strategy for the dynamic and graded control of cell mechanics, motility and matrix remodeling. Biophys. J. 2012, 102, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Pedron, S.; Becka, E.; Harley, B.A. Regulation of glioma cell phenotype in 3d matrices by hyaluronic acid. Biomaterials 2013, 34, 7408–7417. [Google Scholar] [CrossRef] [PubMed]
- Pedron, S.; Becka, E.; Harley, B.A. Spatially gradated hydrogel platform as a 3d engineered tumor microenvironment. Adv. Mater. 2015, 27, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Shivashankar, G.V. Mechanosignaling to the cell nucleus and gene regulation. Annu. Rev. Biophys. 2011, 40, 361–378. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.C.; Whitesides, G.M. Poly(dimethylsiloxane) as a material for fabricating microfluidic devices. Acc. Chem. Res. 2002, 35, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, J.; Mantini, G.; Lu, M.Y.; Fang, H.; Falconi, C.; Chen, L.J.; Wang, Z.L. Quantifying the traction force of a single cell by aligned silicon nanowire array. Nano Lett. 2009, 9, 3575–3580. [Google Scholar] [CrossRef] [PubMed]
- Schoen, I.; Hu, W.; Klotzsch, E.; Vogel, V. Probing cellular traction forces by micropillar arrays: Contribution of substrate warping to pillar deflection. Nano Lett. 2010, 10, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.E.; Su, G.; Pehlke, C.; Trier, S.M.; Eliceiri, K.W.; Keely, P.J.; Friedl, A.; Beebe, D.J. Control of 3-dimensional collagen matrix polymerization for reproducible human mammary fibroblast cell culture in microfluidic devices. Biomaterials 2009, 30, 4833–4841. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Nelson, M.T.; Xue, R.; DeJesus, J.K.; Viapiano, M.S.; Lannutti, J.J.; Sarkar, A.; Winter, J.O. Mimicking white matter tract topography using core-shell electrospun nanofibers to examine migration of malignant brain tumors. Biomaterials 2013, 34, 5181–5190. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Nowicki, M.O.; Lee, C.H.; Chiocca, E.A.; Viapiano, M.S.; Lawler, S.E.; Lannutti, J.J. Quantitative analysis of complex glioma cell migration on electrospun polycaprolactone using time-lapse microscopy. Tissue Eng. Part C 2009, 15, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Rape, A.D.; Kumar, S. A composite hydrogel platform for the dissection of tumor cell migration at tissue interfaces. Biomaterials 2014, 35, 8846–8853. [Google Scholar] [CrossRef] [PubMed]
- Rape, A.D.; Zibinsky, M.; Murthy, N.; Kumar, S. A synthetic hydrogel for the high-throughput study of cell-ecm interactions. Nat. Commun. 2015, 6, 8129. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Irwin, E.F.; Kozhukh, J.; Schaffer, D.V.; Healy, K.E. Biomimetic interfacial interpenetrating polymer networks control neural stem cell behavior. J. Biomed. Mater. Res. A 2007, 81, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, B.; Kim, Y.; Kumar, S. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials 2011, 32, 7913–7923. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Agrawal, B.; Sun, D.; Kuo, J.S.; Williams, J.C. Microfluidics-based devices: New tools for studying cancer and cancer stem cell migration. Biomicrofluidics 2011, 5, 13412. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.J.; Jeon, N.L. Biological applications of microfluidic gradient devices. Integr. Biol. 2010, 2, 584–603. [Google Scholar] [CrossRef] [PubMed]
- Li Jeon, N.; Baskaran, H.; Dertinger, S.K.; Whitesides, G.M.; Van de Water, L.; Toner, M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat. Biotechnol. 2002, 20, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Trappmann, B.; Stapleton, S.C.; Toro, E.; Chen, C.S. Microfluidics embedded within extracellular matrix to define vascular architectures and pattern diffusive gradients. Lab Chip 2013, 13, 3246–3252. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Farach-Carson, M.C.; Jia, X. Three-dimensional in vitro tumor models for cancer research and drug evaluation. Biotechnol. Adv. 2014, 32, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, J.M.; Overstreet, D.J.; Le, L.D.; Vernon, B.L.; Sirianni, R.W. Bioengineered scaffolds for 3d analysis of glioblastoma proliferation and invasion. Ann. Biomed. Eng. 2015, 43, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tong, X.; Yang, F. Bioengineered 3d brain tumor model to elucidate the effects of matrix stiffness on glioblastoma cell behavior using peg-based hydrogels. Mol. Pharm. 2014, 11, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tong, X.; Jiang, X.; Yang, F. Effect of matrix metalloproteinase-mediated matrix degradation on glioblastoma cell behavior in 3d peg-based hydrogels. J. Biomed. Mater. Res. A 2017, 105, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Dejesus, J.; Short, A.R.; Otero, J.J.; Sarkar, A.; Winter, J.O. Glioblastoma behaviors in three-dimensional collagen-hyaluronan composite hydrogels. ACS Appl. Mater. Interfaces 2013, 5, 9276–9284. [Google Scholar] [CrossRef] [PubMed]
- Florczyk, S.J.; Wang, K.; Jana, S.; Wood, D.L.; Sytsma, S.K.; Sham, J.; Kievit, F.M.; Zhang, M. Porous chitosan-hyaluronic acid scaffolds as a mimic of glioblastoma microenvironment ECM. Biomaterials 2013, 34, 10143–10150. [Google Scholar] [CrossRef] [PubMed]
- Kievit, F.M.; Florczyk, S.J.; Leung, M.C.; Wang, K.; Wu, J.D.; Silber, J.R.; Ellenbogen, R.G.; Lee, J.S.; Zhang, M. Proliferation and enrichment of cd133(+) glioblastoma cancer stem cells on 3d chitosan-alginate scaffolds. Biomaterials 2014, 35, 9137–9143. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Kang, S.G.; Kim, P. Strategies of mesenchymal invasion of patient-derived brain tumors: Microenvironmental adaptation. Sci. Rep. 2016, 6, 24912. [Google Scholar] [CrossRef] [PubMed]
- Pedron, S.; Harley, B.A. Impact of the biophysical features of a 3d gelatin microenvironment on glioblastoma malignancy. J. Biomed. Mater. Res. A 2013, 101, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-c stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef] [PubMed]
- Logun, M.T.; Bisel, N.S.; Tanasse, E.A.; Zhao, W.; Gunasekera, B.; Mao, L.; Karumbaiah, L. Glioma cell invasion is significantly enhanced in composite hydrogel matrices composed of chondroitin 4- and 4,6-sulfated glycosaminoglycans. J. Mater. Chem. B Mater. Biol. Med. 2016, 4, 6052–6064. [Google Scholar] [CrossRef] [PubMed]
- Jiguet Jiglaire, C.; Baeza-Kallee, N.; Denicolai, E.; Barets, D.; Metellus, P.; Padovani, L.; Chinot, O.; Figarella-Branger, D.; Fernandez, C. Ex vivo cultures of glioblastoma in three-dimensional hydrogel maintain the original tumor growth behavior and are suitable for preclinical drug and radiation sensitivity screening. Exp. Cell Res. 2014, 321, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Coquerel, B.; Poyer, F.; Torossian, F.; Dulong, V.; Bellon, G.; Dubus, I.; Reber, A.; Vannier, J.P. Elastin-derived peptides: Matrikines critical for glioblastoma cell aggressiveness in a 3-d system. Glia 2009, 57, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Guo, T.W.; Wu, A.P.; Wells, A.; Gertler, F.B.; Lauffenburger, D.A. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3d arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol. Biol. Cell 2008, 19, 4249–4259. [Google Scholar] [CrossRef] [PubMed]
- Nie, T.; Baldwin, A.; Yamaguchi, N.; Kiick, K.L. Production of heparin-functionalized hydrogels for the development of responsive and controlled growth factor delivery systems. J. Control. Release 2007, 122, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; Jain, A.; Tanner, K.; MacKay, J.L.; Kumar, S. Probing cellular mechanobiology in three-dimensional culture with collagen-agarose matrices. Biomaterials 2010, 31, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Vunjak-Novakovic, G. Modeling tumor microenvironments using custom-designed biomaterial scaffolds. Curr. Opin. Chem. Eng. 2016, 11, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Bogorad, M.I.; DeStefano, J.; Karlsson, J.; Wong, A.D.; Gerecht, S.; Searson, P.C. Review: In vitro microvessel models. Lab Chip 2015, 15, 4242–4255. [Google Scholar] [CrossRef] [PubMed]
- Bogorad, M.I.; DeStefano, J.; Wong, A.D.; Searson, P.C. Tissue-engineered 3d microvessel and capillary network models for the study of vascular phenomena. Microcirculation 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Chonan, Y.; Taki, S.; Sampetrean, O.; Saya, H.; Sudo, R. Endothelium-induced three-dimensional invasion of heterogeneous glioma initiating cells in a microfluidic coculture platform. Integr. Biol. 2017, 9, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Pei, Y.; Xie, M.; Jin, Z.H.; Xiao, Y.S.; Wang, Y.; Zhang, L.N.; Li, Y.; Huang, W.H. An artificial blood vessel implanted three-dimensional microsystem for modeling transvascular migration of tumor cells. Lab Chip 2015, 15, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Searson, P.C. Live-cell imaging of invasion and intravasation in an artificial microvessel platform. Cancer Res. 2014, 74, 4937–4945. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, K.M.; Potter, D.R.; Tien, J. Formation of perfused, functional microvascular tubes in vitro. Microvasc. Res. 2006, 71, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Stevens, K.R.; Yang, M.T.; Baker, B.M.; Nguyen, D.-H.T.; Cohen, D.M.; Toro, E.; Chen, A.A.; Galie, P.A.; Yu, X.; et al. Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues. Nat. Mater. 2012, 11, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Qazi, H.; Shi, Z.-D.; Tarbell, J.M. Fluid shear stress regulates the invasive potential of glioma cells via modulation of migratory activity and matrix metalloproteinase expression. PLoS ONE 2011, 6, e20348. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Yan, T.; Zhu, H.; Liang, X.; Leiss, L.; Sakariassen, P.O.; Skaftnesmo, K.O.; Huang, B.; Costea, D.E.; Enger, P.O.; et al. A co-culture model with brain tumor-specific bioluminescence demonstrates astrocyte-induced drug resistance in glioblastoma. J. Transl. Med. 2014, 12, 278. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.; Miller, I.; Symons, M.; Segall, J.E. Coculture assays to study macrophage and microglia stimulation of glioblastoma invasion. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, B.D.; Mui, K.L.; Driscoll, T.P.; Caliari, S.R.; Mehta, K.D.; Assoian, R.K.; Burdick, J.A.; Mauck, R.L. N-cadherin adhesive interactions modulate matrix mechanosensing and fate commitment of mesenchymal stem cells. Nat. Mater. 2016, 15, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in BFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhang, Q.B.; Dong, J.; Wu, Y.Y.; Shen, Y.T.; Zhao, Y.D.; Zhu, Y.D.; Diao, Y.; Wang, A.D.; Lan, Q. Glioma stem cells are more aggressive in recurrent tumors with malignant progression than in the primary tumor and both can be maintained long-term in vitro. BMC Cancer 2008, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Potter, N.E.; Phipps, K.; Harkness, W.; Hayward, R.; Thompson, D.; Jacques, T.S.; Harding, B.; Thomas, D.G.; Rees, J.; Darling, J.L.; et al. Astrocytoma derived short-term cell cultures retain molecular signatures characteristic of the tumour in situ. Exp. Cell Res. 2009, 315, 2835–2846. [Google Scholar] [CrossRef] [PubMed]
- Lewandowicz, G.M.; Harding, B.; Harkness, W.; Hayward, R.; Thomas, D.G.; Darling, J.L. Chemosensitivity in childhood brain tumours in vitro: Evidence of differential sensitivity to lomustine (ccnu) and vincristine. Eur. J. Cancer 2000, 36, 1955–1964. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Zhang, S.; Xie, R.; Gao, B.; Campos, B.; Herold-Mende, C.; Lei, T. The utility and limitations of neurosphere assay, cd133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010, 20, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Fael Al-Mayhani, T.M.; Ball, S.L.; Zhao, J.W.; Fawcett, J.; Ichimura, K.; Collins, P.V.; Watts, C. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J. Neurosci. Methods 2009, 176, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Shaw, A.K.; Strand, D.W.; Hayward, S.W. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 2010, 21, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- LeBeau, A.M.; Brennen, W.N.; Aggarwal, S.; Denmeade, S.R. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol. Cancer Ther. 2009, 8, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.A.; Varro, A.; Wang, T.C.; Tycko, B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Semin. Cell Dev. Biol. 2010, 21, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Buffo, A.; Rite, I.; Tripathi, P.; Lepier, A.; Colak, D.; Horn, A.P.; Mori, T.; Gotz, M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl. Acad. Sci. USA 2008, 105, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Berninger, B.; Gotz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Fomchenko, E.I.; Dougherty, J.D.; Helmy, K.Y.; Katz, A.M.; Pietras, A.; Brennan, C.; Huse, J.T.; Milosevic, A.; Holland, E.C. Recruited cells can become transformed and overtake PDGF-induced murine gliomas in vivo during tumor progression. PLoS ONE 2011, 6, e20605. [Google Scholar] [CrossRef] [PubMed]
- Cesselli, D.; Beltrami, A.P.; Pucer, A.; Bourkoula, E.; Ius, T.; Vindigni, M.; Skrap, M.; Beltrami, C.A. Human low grade glioma cultures. In Diffuse Low-Grade Gliomas in Adults; Duffau, H., Ed.; Springer: London, UK, 2013; pp. 137–163. [Google Scholar]
- Caponnetto, F.; Beltrami, A.P.; Ius, T.; Skrap, M.; Cesselli, D. Diffule low-grade glioma associated stem cells. In Diffuse Low-Grade Gliomas in Adults; Duffau, H., Ed.; Springer International Publishing: London, UK, 2017; pp. 151–172. [Google Scholar]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from glioma-associated mesenchymal stem cells increase the tumorigenicity of glioma stem-like cells via transfer of mir-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, L.; Bourkoula, E.; Migliorini, E.; Palma, A.; Pucer, A.; Skrap, M.; Scoles, G.; Beltrami, A.P.; Cesselli, D.; Lazzarino, M. Investigation of adhesion and mechanical properties of human glioma cells by single cell force spectroscopy and atomic force microscopy. PLoS ONE 2014, 9, e112582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ius, T.; Ciani, Y.; Ruaro, M.E.; Isola, M.; Sorrentino, M.; Bulfoni, M.; Candotti, V.; Correcig, C.; Bourkoula, E.; Manini, I.; et al. A nf-kappab signature predicts low-grade glioma prognosis: A precision medicine approach based on patient-derived stem cells. Neuro Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.; Bonner, M.; Fried, L.; Hofmekler, J.; Arbiser, J.; Bellamkonda, R. Identifying new small molecule anti-invasive compounds for glioma treatment. Cell Cycle 2013, 12, 2200–2209. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated mgmt promoter (centric eortc 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of pi3k inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011, 13, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.M.; Demuth, T.; Mobley, D.; Berens, M.; Sander, L.M. A mathematical model of glioblastoma tumor spheroid invasion in a three-dimensional in vitro experiment. Biophys. J. 2007, 92, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.R.; Sontheimer, H. Inhibition of the sodium-potassium-chloride cotransporter isoform-1 reduces glioma invasion. Cancer Res. 2010, 70, 5597–5606. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Michiue, H.; Yamada, H.; Takata, K.; Nakayama, H.; Wei, F.Y.; Fujimura, A.; Tazawa, H.; Asai, A.; Ogo, N.; et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Sci. Rep. 2016, 6, 23372. [Google Scholar] [CrossRef] [PubMed]
- Chikano, Y.; Domoto, T.; Furuta, T.; Sabit, H.; Kitano-Tamura, A.; Pyko, I.V.; Takino, T.; Sai, Y.; Hayashi, Y.; Sato, H.; et al. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, Rac1 and c-Jun N-terminal kinase-mediated pathway. Mol. Cancer Ther. 2015, 14, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.L.; Parat, M.O.; Binder, Z.A.; Gallia, G.L.; Riggins, G.J. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration and invasion in glioblastoma multiforme cells. Oncotarget 2011, 2, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yoshida, D.; Liu, S.; Teramoto, A. Inhibition of cell invasion by indomethacin on glioma cell lines: In vitro study. J. Neurooncol. 2005, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Kijewska, M.; Lipko, M.; Hibner, U.; Kaminska, B. Downregulation of Akt and FAK phosphorylation reduces invasion of glioblastoma cells by impairment of mt1-mmp shuttling to lamellipodia and downregulates mmps expression. Biochim. Biophys. Acta 2011, 1813, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Tritschler, I.; Adams, B.; Tabatabai, G.; Wick, W.; Stupp, R.; Weller, M. Cilengitide modulates attachment and viability of human glioma cells but not sensitivity to irradiation or temozolomide in vitro. Neuro Oncol. 2009, 11, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, V.C.; Lung, S.S.; Pu, J.K.; Hung, K.N.; Leung, G.K. Invasion of human glioma cells is regulated by multiple chloride channels including clc-3. Anticancer Res. 2010, 30, 4515–4524. [Google Scholar] [PubMed]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Modulation of glioma cell migration and invasion using Cl(−) and K(+) ion channel blockers. J. Neurosci. 1999, 19, 5942–5954. [Google Scholar] [PubMed]
- Westhoff, M.A.; Zhou, S.; Nonnenmacher, L.; Karpel-Massler, G.; Jennewein, C.; Schneider, M.; Halatsch, M.E.; Carragher, N.O.; Baumann, B.; Krause, A.; et al. Inhibition of nf-kappab signaling ablates the invasive phenotype of glioblastoma. Mol. Cancer Res. 2013, 11, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Fried, L.; Rowson, S.A.; Bonner, M.Y.; Karumbaiah, L.; Diaz, B.; Courtneidge, S.A.; Knaus, U.G.; Brat, D.J.; Arbiser, J.L.; et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 2012, 4, 127ra36. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.; Esteves, F.; Chakrabarty, A.; Cockle, J.; Short, S.; Bruning-Richardson, A. High-content analysis of tumour cell invasion in three-dimensional spheroid assays. Oncoscience 2015, 2, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Terzis, A.J.; Fiskerstrand, T.; Refsum, H.; Ueland, P.M.; Arnold, H.; Bjerkvig, R. Proliferation, migration and invasion of human glioma cells exposed to antifolate drugs. Int. J. Cancer 1993, 54, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Lakka, S.S.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Downregulation of upa, upar and mmp-9 using small, interfering, hairpin rna (sirna) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004, 1, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin b and mmp-9 gene expression in glioblastoma cell line via rna interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681–4689. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Mechanism Studied | Significance | Reference |
|---|---|---|---|
| 2-D Models | |||
| Monolayer culture on glass or plastic slides (scratch assays) | Cell motility | Used to define the effect of several ECM components and soluble factors on glioma cell motility. | [35,36,37,38,39,40,41,42,43,44,45,46,47,48,103,104,105,113] |
| Transwell migration | Cell motility and cell invasion depending on: | ||
| Chemotactic gradient | Used to define factors able to favour or inhibit glioma invasion. | [115,116,117,118] | |
| Insert coating | Used to assess the role of ECM components on cell invasion. | [40,58,113] | |
| Pore size (e.g., 8 μm vs. 3 μm) | Glioma cells requires myosin II only when migrating through 3 μm in diameter pores. | [14] | |
| 3D Models | |||
| Modified transwell migration | Trans-endothelial migration assay: effect of endothelial cells on glioma invasion | Role of bradykinin in the perivascular invasion. | [13,125] |
| Brain slice invasion assay: invasion of brain tissue slices | Effects of ECM components, soluble factors and drugs on glioma cell motility. | [126,127,128] | |
| Spheroids | Multicellular tumour spheroids (MTS) | Effects of motogenic substances and irradiation on migration upon adhesion on plastic substrates. | [134,135,136,137] |
| Organotypic multicellular spheroids (OMS) | Role of different ECM components on cell migration from patient derived spheroids. | [133,138,139] | |
| Ex vivo tumour sections | Tumour slices of PDGF-driven rat gliomas: glioma migration in living brain tissue through extracellular spaces that are in the submicrometer range | When invading the extracellular spaces, glioma cells squeeze through pores smaller than their nuclear diameter and this process requires myosin II. | [14] |
| Tumour slices of brains xenotransplanted with human tumour cells: perivascular invasion of glioma cells | Perivascular glioma cells disrupt both astrocyte–vascular coupling and the blood–brain barrier. | [55] | |
| Engineered Models | |||
| 2D | Stiffness: substrata with controlled elastic modulus | An increase in ECM stiffness induces motility of glioma cells. Mechanotransduction involves non-muscle myosin II, α-actinin, talin and Rho GTPase RhoA. | [51,150,151,152] |
| Physical topography and confinement of cells | Cells cultured in micron-sized channels or on substrates with aligned nanofibers showed an increased motility. | [146,157,158,159,160,161] | |
| ECM composition and chemotactic gradients | Definition of the role of ECM components (e.g., integrins and CD44/HA) and chemotactic gradient on tumour cell motility. | [147,162,163,164,165,166,167,168] | |
| 3D | Stiffness | Cell motility resulted to be inversely related to the stiffness. However, regarding MMP secretion, HA-matrices resulted to either enhance or decrease MMP9 secretion. | [153,154,165,171,172] |
| ECM composition | Role of different ECM-components, utilized to construct 3D hydrogels, such as HA, chondroitin sulphate, chitosan and collagen/gelatine, on migration of commercially available and patient-derived GBM cell lines. Evaluation of the underlined pathways (e.g., production of matrix degrading enzymes such as hyaluronidases, MMPs and HIF). Evaluation of the pro-migratory effect of EGF and the role played by heparin-cytokines interaction. | [153,154,162,165,173,174,175,176,177,178,179,180,181,183,184] | |
| Migration along constrained paths | Mechanisms underlying the parenchyma invasion. | [154,163,176,179,185] | |
| Perivascular invasion: 3D models available but not yet tested with glioma cells. | |||
| Interstitial flow | Contrasting results showing the pro-migratory and anti-migratory effects of the interstitial flow. Role of CD44-mediated mechanotransduction and autologous chemotaxis via CXCR4–CXCL12 signalling. | [59,195] | |
| 3D cell-cell interaction | Anti-apoptotic effect of astrocytes co-cultured with tumour cells. | [196] | |
| Pro-migratory effects of microglial cells co-cultured with tumour cells. | [197] | ||
| Model | Drugs | Mechanism of Action | Reference |
|---|---|---|---|
| 2-D Models | |||
| Scratch assay/Transwell migration | Bumetanide | The inhibition of the Sodium-Potassium-Chloride Cotransporter Isoform-1 (NKCC1) affect cell motility only when cells had to undergo volume changes during migration. | [231] |
| Fluvoxamine | Selective serotonin reuptake inhibitor (SSRI) disrupting actin polymerization and inhibiting glioma motility and invasion. | [232] | |
| Glycogen synthase kinase-3 β (GSK-3β) inhibitors | Glycogen synthase kinase (GSK) 3beta inhibitors are able to attenuate glioma invasion in vitro and in vivo. | [233] | |
| Blebbistatin and Rho-associated kinase (ROCK) inhibitor Y-27632 | Inhibitors of non-muscle myosin (NMMII) IIA and IIB affect glioma cells migration through 3 μm in diameter pores (confined spaces). | [14] | |
| PIK3CA or PIK3R1 abrogation by lentiviral-mediated shRNA | Abrogation of PIK3CA or PIK3R1 reduces glioma invasion and motility in vitro. | [234] | |
| Sulfasalazine | The block of the system xc inhibit glutamate release thus reducing chemotactic invasion and scrape motility assays. | [235] | |
| HIF-1α abrogation by lentiviral-mediated shRNA | Knock down of HIF-1α in glioma cells significantly impairs their migration in vitro and in vivo. | [64] | |
| Indomethacin | This non-steroidal anti-inflammatory drug is able to reduce glioma cell invasion mediated by MMP-2 and MMP-9. | [236] | |
| Cyclosporin A (CsA) | CsA impairs migration and invasion of human glioblastoma cells by downregulation of Akt phosphorylation. | [237] | |
| Cilengitide | Integrin (αvβ3 and αvβ5) inhibitor able to reduce glioma invasion in vitro. | [238] | |
| Cholorotoxin | The inhibitor of the chloride channel-3 (ClC-3) partly inhibits glioma migration by disrupting volume changes. | [239,240] | |
| Disulfiram | NF-kB inhibitor able to reduce glioma cell invasion. | [241] | |
| Imipramine Blue | It inhibits NADPH oxidase-mediated reactive oxygen species generation and modifies the expression of actin regulatory elements reducing glioma invasion in vitro and in vivo. | [242] | |
| 3D Models | |||
| Modified transwell migration | Icatibant | B2 bradykinin receptor inhibitor acting on the perivascular invasion. | [13] |
| Autocamtide-2 related inhibitory peptide (AIP) | CaMKII (Ca2+/calmodulin-dependent protein kinase II) inhibitor are involved in the hydrodynamic model of cell invasion. | [125] | |
| Vincristin and paclitaxel | Cytoskeleton destabilizers are able to inhibit glioma cell invasion in a dose dependent manner. | [126] | |
| Spheroids | Antibodies to the EGF receptor | Effects of antibodies on proliferation and migration in multicellular tumour spheroids. | [135] |
| Lithium chloride (LiCl) and Bio-Indirubin (BIO) | Glycogen synthase kinase-3 β (GSK-3β) inhibitors are able to inhibit cell invasion in multicellular tumour spheroids. | [243] | |
| Methotrexate (MTX) and trimetrexate (TMX) | Anti-folate agents affect glioma invasion in 2D culture but not in multicellular tumour spheroids. | [244] | |
| Downregulation of cathepsin B, uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) | Retardation of glioma cell invasion in vitro and in vivo. | [245,246] | |
| Ex vivo tumour sections | Blebbistatin and Rho-associated kinase (ROCK) inhibitor Y-27632 | Inhibitors of NMMIIA and IIB are involved in glioma cells migration in living brain tissue through confined spaces. | [14] |
| Engineered Models | |||
| 2D | Blebbistatin and Rho-associated kinase (ROCK) inhibitor Y-27632 | Pharmacological inhibition of NMMII or its upstream regulator ROCK blunts the sensitivity of glioma cells to ECM stiffness and renders this relationship insensitive to matrix confinement. | [51,146] |
| Suppression of α-actinin-1 and α-actinin-4 by small interfering RNA (siRNA) | Disruption of α-actinin-1 and α-actinin-4 reduces cell motility and the sensitivity of glioma cells to ECM stiffness | [150] | |
| Tyrphostin Triciribine Wortmannin | Stiffness-dependent glioma cell behaviour is altered by treatment with EGFR inhibitor (Tyrphostin), Akt inhibitor (Triciribine) and PI3 Kinase inhibitor (Wortmannin). | [147] | |
| 3D | GM6001 BB94 TIMP1 MMP12 function blocking antibody | The tenascin-C–mediated invasiveness can be blocked by broad-spectrum metalloproteinase inhibitors (GM6001 and BB94). However, this effect did not involve MMP-2 and MMP-9, as shown in 2D assays but MMP12. | [179] |
| GM6001 | The broad-spectrum MMP inhibitor is able to interfere with the EGF-induced glioma cell migration in 3D. | [183] | |
| The broad-spectrum MMP inhibitor is able to interfere with the flow-modulated motility. | [195] | ||
| AMD 3100 | Non-competitive CXCR4 inhibitor interfering with the interstitial flow enhanced invasion. | [58] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010147
Manini I, Caponnetto F, Bartolini A, Ius T, Mariuzzi L, Di Loreto C, Beltrami AP, Cesselli D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. International Journal of Molecular Sciences. 2018; 19(1):147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010147
Chicago/Turabian StyleManini, Ivana, Federica Caponnetto, Anna Bartolini, Tamara Ius, Laura Mariuzzi, Carla Di Loreto, Antonio Paolo Beltrami, and Daniela Cesselli. 2018. "Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models" International Journal of Molecular Sciences 19, no. 1: 147. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010147