The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree


Abstract
:1. Introduction
2. Results and Discussion
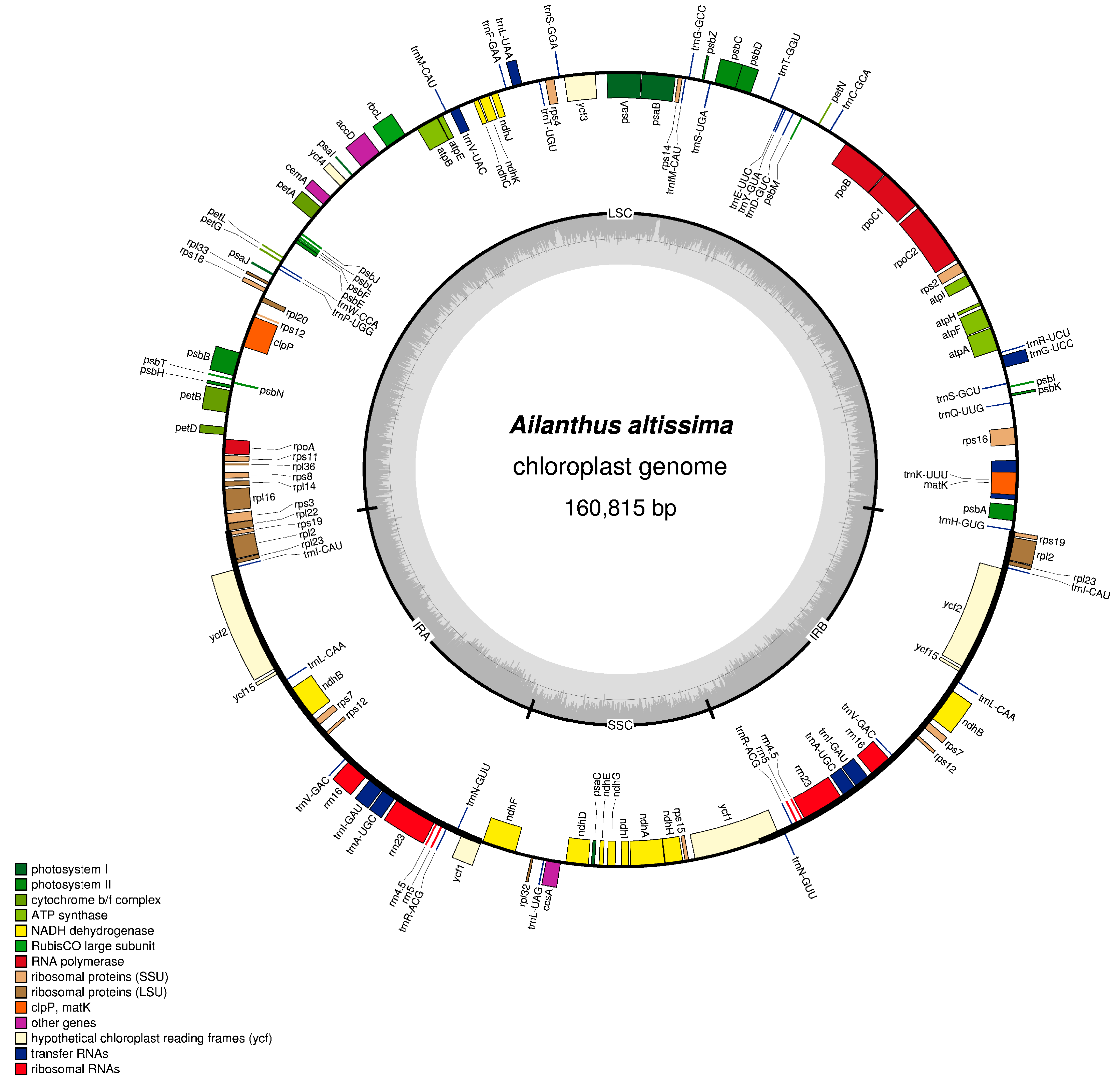
2.1. Ailanthus altissima Genome Size and Features
2.2. IR Expansion and Contraction and Genome Rearrangement
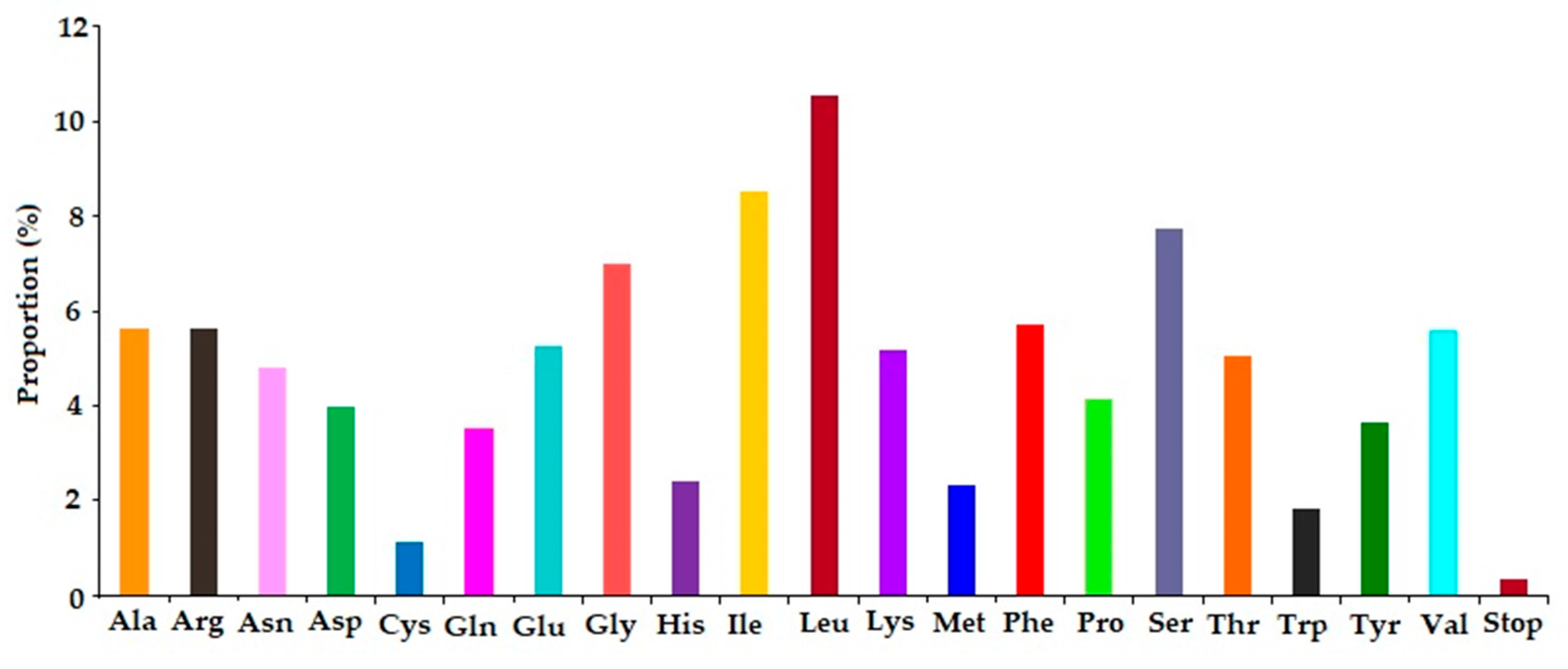
2.3. Codon Usage and Putative RNA Editing Sites in Chloroplast Genes of A. altissima
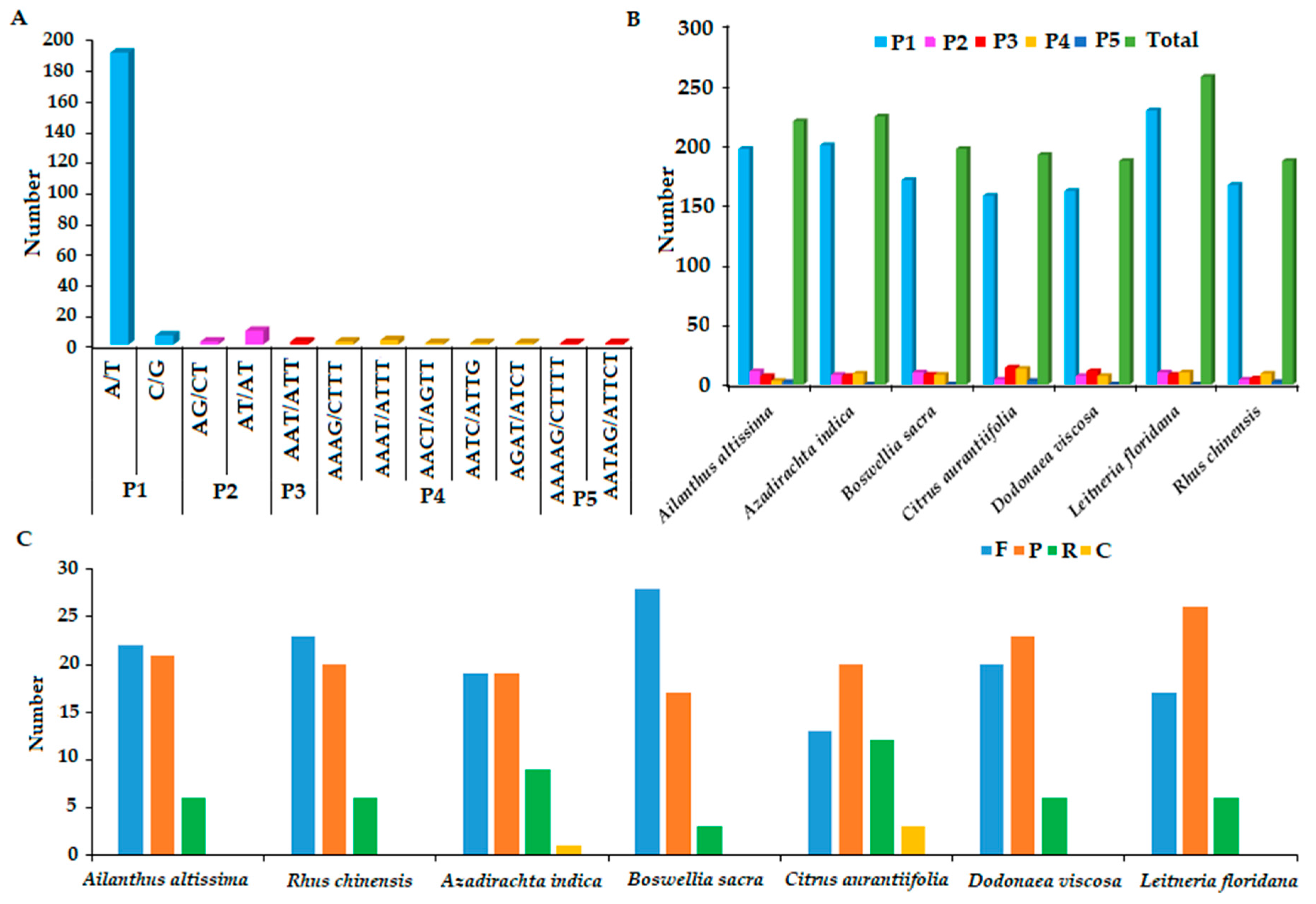
2.4. Repeat Sequence Analysis
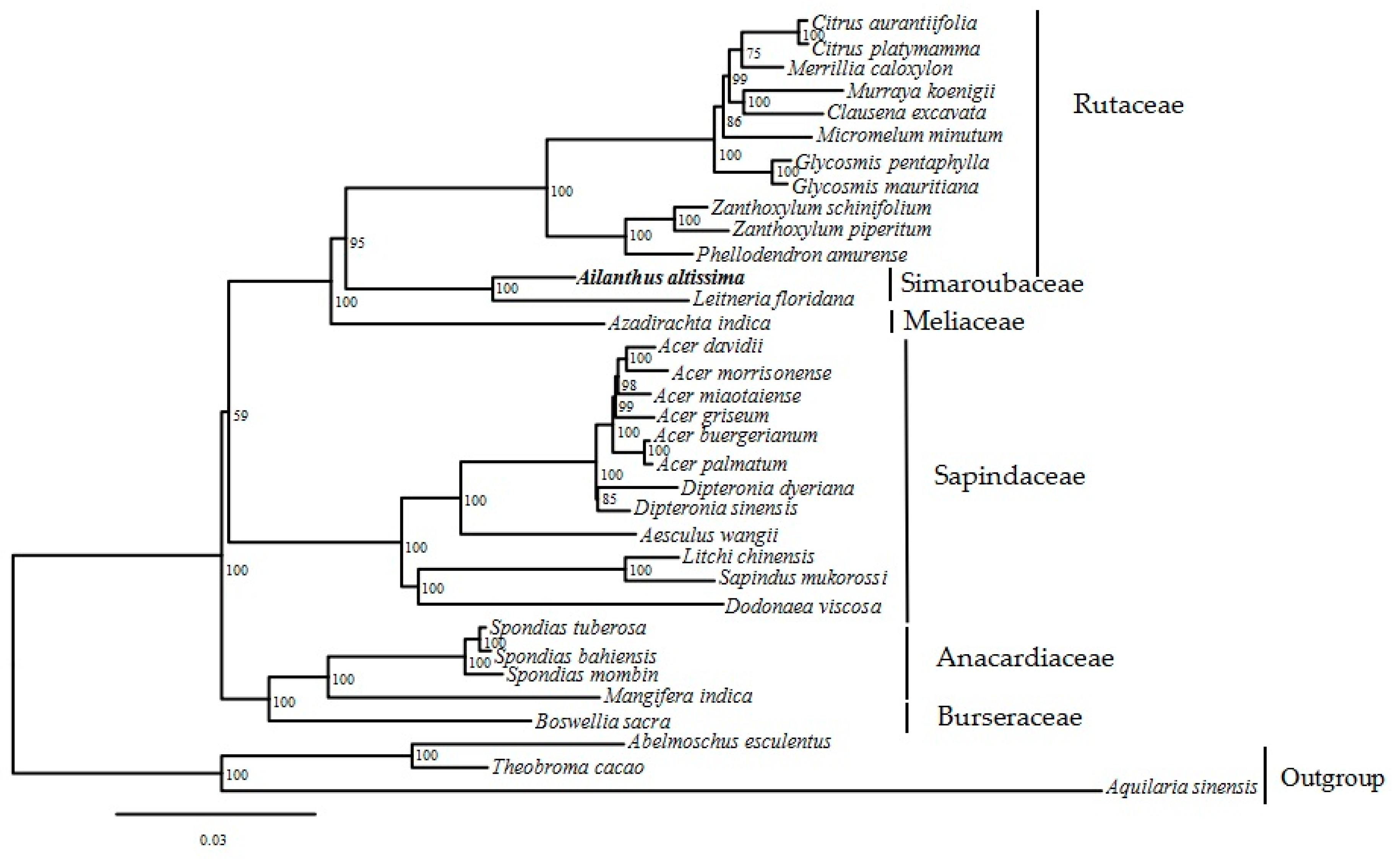
2.5. Phylogenetic Tree
3. Materials and Methods
3.1. Plant Materials and DNA Extraction
3.2. The Tree of Heaven Plastome Sequence Assembly and Annotation
3.3. Genome Comparison and Gene Rearrangement
3.4. Repeat Analysis in A. altissima Chloroplast Genome
3.5. Codon Usage and RNA Editing Sites
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SC | Single copy |
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
References
- Kowarik, I.; Säumel, I. Biological flora of central Europe: Ailanthus altissima (Mill.) swingle. Perspect. Plant Ecol. Evol. Syst. 2007, 8, 207–237. [Google Scholar] [CrossRef]
- Liao, Y.Y.; Guo, Y.H.; Chen, J.M.; Wang, Q.F. Phylogeography of the widespread plant Ailanthus altissima (Simaroubaceae) in China indicated by three chloroplast DNA regions. J. Syst. Evol. 2014, 52, 175–185. [Google Scholar] [CrossRef]
- Kurokochi, H.; Saito, Y.; Ide, Y. Genetic structure of the introduced heaven tree (Ailanthus altissima) in Japan: Evidence for two distinct origins with limited admixture. Botany 2014, 93, 133–139. [Google Scholar] [CrossRef]
- Dallas, J.F.; Leitch, M.J.; Hulme, P.E. Microsatellites for tree of heaven (Ailanthus altissima). Mol. Ecol. Resour. 2005, 5, 340–342. [Google Scholar] [CrossRef]
- Aldrich, P.R.; Briguglio, J.S.; Kapadia, S.N.; Morker, M.U.; Rawal, A.; Kalra, P.; Huebner, C.D.; Greer, G.K. Genetic structure of the invasive tree Ailanthus altissima in eastern United States cities. J. Bot. 2010, 2010, 795735. [Google Scholar]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J. Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Cui, L.; Moret, B.M.; Tang, J. Gene rearrangement analysis and ancestral order inference from chloroplast genomes with inverted repeat. BMC Genom. 2008, 9, S25. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.-B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogen. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.; Jansen, R.K.; Chumley, T.W.; Kim, K.-J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.-J.; Wang, T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Wang, Y.-N.; Hsu, C.-Y.; Lin, C.-P.; Chaw, S.-M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and Cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Saski, C.; Lee, S.-B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2010, 28, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Steane, D.A. Complete nucleotide sequence of the chloroplast genome from the tasmanian blue gum, Eucalyptus globulus (myrtaceae). DNA Res. 2005, 12, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Lin, C.-P.; Hsu, C.-Y.; Wang, R.-J.; Chaw, S.-M. Comparative chloroplast genomes of Pinaceae: Insights into the mechanism of diversified genomic organizations. Genome Biol. Evol. 2011, 3, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, E.; Gemmell, N.J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, Y.D.; Tyekucheva, S.; Chiaromonte, F.; Makova, K.D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008, 18, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Peakall, R. A new set of universal de novo sequencing primers for extensive coverage of noncoding chloroplast DNA: New opportunities for phylogenetic studies and CPSSR discovery. Mol. Ecol. Resour. 2009, 9, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Watanabe, A.; Miyamoto, N.; Takata, K. Development and characterization of chloroplast microsatellite markers for Cryptomeria japonica D. Don. Mol. Ecol. Resour. 2009, 9, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento Vieira, L.; Faoro, H.; Rogalski, M.; de Freitas Fraga, H.P.; Cardoso, R.L.A.; de Souza, E.M.; de Oliveira Pedrosa, F.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-J.; Hogenhout, S.A.; Al-Sadi, A.M.; Kuo, C.-H. Complete chloroplast genome sequence of Omani lime (Citrus aurantiifolia) and comparative analysis within the rosids. PLoS ONE 2014, 9, e113049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Park, J.; Lee, H.; Sohn, S.-H.; Lee, J. Complete chloroplast genomic sequence of Citrus platymamma determined by combined analysis of sanger and NGS data. Hortic. Environ. Biotechnol. 2015, 56, 704–711. [Google Scholar] [CrossRef]
- Saina, J.K.; Gichira, A.W.; Li, Z.-Z.; Hu, G.-W.; Wang, Q.-F.; Liao, K. The complete chloroplast genome sequence of Dodonaea viscosa: Comparative and phylogenetic analyses. Genetica 2018, 146, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, S. The complete chloroplast genome sequence of Ampelopsis: Gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 2016, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Kim, W.J.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis maxim. In addition, Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Complete chloroplast genome sequence of Decaisnea insignis: Genome organization, genomic resources and comparative analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, H.-L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.L.; Al-Harrasi, A.; Asaf, S.; Park, C.E.; Park, G.-S.; Khan, A.R.; Lee, I.-J.; Al-Rawahi, A.; Shin, J.-H. The first chloroplast genome sequence of Boswellia sacra, a resin-producing plant in Oman. PLoS ONE 2017, 12, e0169794. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yue, M.; Niu, C.; Ma, X.-F.; Li, Z.-H. Comparative analysis of the complete chloroplast genome of four endangered herbals of Notopterygium. Genes 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Guo, L.; Zhao, W.; Xu, J.; Li, Y.; Zhang, X.; Shen, X.; Wu, M.; Hou, X. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules 2018, 23, 246. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Z.; Saina, J.K.; Gichira, A.W.; Kyalo, C.M.; Wang, Q.-F.; Chen, J.-M. Comparative genomics of the Balsaminaceae sister genera Hydrocera triflora and Impatiens pinfanensis. Int. J. Mol. Sci. 2018, 19, 319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y.; Song, J.; Yao, H. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, Z.; Zhang, T.; Zhong, W.; Liu, C.; Yuan, Q.; Huang, L. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in Scutellaria. Genes 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cui, Y.; Chen, X.; Li, Y.; Xu, Z.; Duan, B.; Li, Y.; Song, J.; Yao, H. Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: Molecular structures, comparative analysis, and phylogenetic analysis. Molecules 2018, 23, 437. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zheng, Y.; Liu, S.; Zhong, Y.; Wu, Y.; Li, J.; Xu, L.-A.; Xu, M. The complete chloroplast genome of Cinnamomum camphora and its comparison with related Lauraceae species. PeerJ 2017, 5, e3820. [Google Scholar] [CrossRef] [PubMed]
- De Santana Lopes, A.; Pacheco, T.G.; Nimz, T.; do Nascimento Vieira, L.; Guerra, M.P.; Nodari, R.O.; de Souza, E.M.; de Oliveira Pedrosa, F.; Rogalski, M. The complete plastome of macaw palm [Acrocomia aculeata (jacq.) lodd. Ex mart.] and extensive molecular analyses of the evolution of plastid genes in arecaceae. Planta 2018, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, F.; Nie, X.; Xing, G.; Zhao, X.; Lin, Y.; Wang, S.; Weining, S. Identification and characterisation of rna editing sites in chloroplast transcripts of einkorn wheat (Triticum monococcum). Ann. Appl. Biol. 2018, 172, 197–207. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Cho, S.-T.; Haryono, M.; Kuo, C.-H. Complete chloroplast genome sequence of common Bermuda grass (Cynodon dactylon (L.) pers.) and comparative analysis within the family poaceae. PLoS ONE 2017, 12, e0179055. [Google Scholar]
- Park, M.; Park, H.; Lee, H.; Lee, B.-H.; Lee, J. The complete plastome sequence of an antarctic bryophyte Sanionia uncinata (Hedw.) loeske. Int. J. Mol. Sci. 2018, 19, 709. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J. RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef] [PubMed]
- De Santana Lopes, A.; Pacheco, T.G.; dos Santos, K.G.; do Nascimento Vieira, L.; Guerra, M.P.; Nodari, R.O.; de Souza, E.M.; de Oliveira Pedrosa, F.; Rogalski, M. The Linum usitatissimum L. Plastome reveals atypical structural evolution, new editing sites, and the phylogenetic position of Linaceae within malpighiales. Plant Cell Rep. 2018, 37, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.W.; Fernando, E.S.; Soltis, P.S.; Soltis, D.E. Molecular phylogeny of the tree-of-heaven family (Simaroubaceae) based on chloroplast and nuclear markers. Int. J. Plant Sci. 2007, 168, 1325–1339. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, I.; Kim, J.-K.; Park, J.Y.; Joh, H.J.; Park, H.-S.; Lee, H.O.; Lee, S.-C.; Hur, Y.-J.; Yang, T.-J. The complete chloroplast genome sequence of Rhus chinensis mill (Anacardiaceae). Mitochondrial DNA Part B 2016, 1, 696–697. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The prep suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Category | Group of Genes | Gene Name | Number |
|---|---|---|---|
| Self-replication | rRNA genes | rrn16(×2), rrn23(×2), rrn4.5(×2), rrn5(×2), | 4 |
| tRNA genes | trnA-UGC*(×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC, trnH-GUG, trnI-CAU(×2), trnI-GAU*(×2), trnK-UUU*, trnL-CAA(×2), trnL-UAA*, trnL-UAG, trnG-GCC*, trnM-CAU, trnN-GUU(×2), trnP-GGG, trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC*, trnW-CCA, trnY-GUA | 30 | |
| Ribosomal small subunit | rps2, rps3, rps4, rps7(×2), rps8, rps11, rps12, rps14, rps15, rps16*, rps18, rps19 | 12 | |
| Ribosomal large subunit | rpl2*(×2), rpl14, rpl16, rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 | 9 | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 | 4 | |
| Photosynthesis | Large subunit of rubisco | rbcL | 1 |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf3** | 6 | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 | |
| NADH dehydrogenase | ndhA*, ndhB*(×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 11 | |
| Cytochrome b/f complex | petA, petB*, petD, petG, petL, petN | 6 | |
| ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | 6 | |
| Other | Maturase | matK | 1 |
| Subunit of acetyl-CoA carboxylase | accD | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Protease | clpP** | 1 | |
| c-type cytochrome synthesis | ccsA | 1 | |
| Functions unknown | Conserved open reading frames (ycf) | ycf1, ycf2(×2), ycf4, ycf15(×2) | 4 |
| Total | 113 |
| Gene | Nucleotide Position | Amino Acid Position | Codon Conversion | Amino Acid Conversion | Score |
|---|---|---|---|---|---|
| accD | 818 | 273 | TCG ≥ TTG | S ≥ L | 0.80 |
| atpF | 92 | 31 | CCA ≥ CTA | P ≥ L | 0.86 |
| 353 | 118 | TCA ≥ TTA | S ≥ L | 1.00 | |
| atpB | 403 | 135 | CCA ≥ TCA | P ≥ S | 0.86 |
| rps14 | 80 | 27 | TCA ≥ TTA | S ≥ L | 1.00 |
| 149 | 50 | TCA ≥ TTA | S ≥ L | 1.00 | |
| ccsA | 145 | 49 | CTT ≥ TTT | L ≥ F | 1.00 |
| clpP | 556 | 186 | CAT ≥ TAT | H ≥ Y | 1.00 |
| MatK | 319 | 107 | CTT ≥ TTT | L ≥ F | 0.86 |
| 457 | 153 | CAC ≥ TAC | H ≥ Y | 1.00 | |
| 643 | 215 | CAT ≥ TAT | H ≥ Y | 1.00 | |
| 1246 | 416 | CAC ≥ TAC | H ≥ Y | 1.00 | |
| ndhA | 107 | 36 | CCT ≥ CTT | P ≥ L | 1.00 |
| 341 | 114 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 566 | 189 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 1073 | 358 | TCC ≥ TTC | S ≥ F | 1.00 | |
| ndhB | 149 | 50 | TCA ≥ TTA | S ≥ L | 1.00 |
| 467 | 156 | CCA ≥ CTA | P ≥ L | 1.00 | |
| 586 | 196 | CAT ≥ TAT | H ≥ Y | 1.00 | |
| 611 | 204 | TCA ≥ TTA | S ≥ L | 0.80 | |
| 746 | 249 | TCT ≥ TTT | S ≥ F | 1.00 | |
| 830 | 277 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 836 | 279 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 1255 | 419 | CAT ≥ TAT | H ≥ Y | 1.00 | |
| 1481 | 494 | CCA ≥ CTA | P ≥ L | 1.00 | |
| ndhD | 2 | 1 | ACG ≥ ATG | T ≥ M | 1.00 |
| 313 | 105 | CGG ≥ TGG | R ≥ W | 0.80 | |
| 383 | 128 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 674 | 225 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 878 | 293 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 887 | 296 | CCT ≥ CTT | P ≥ L | 1.00 | |
| 1076 | 359 | GCT ≥ GTT | A ≥ V | 1.00 | |
| 1298 | 433 | TCA ≥ TTA | S ≥ L | 0.80 | |
| 1310 | 437 | TCA ≥ TTA | S ≥ L | 0.80 | |
| ndhF | 290 | 97 | TCA ≥ TTA | S ≥ L | 1.00 |
| 586 | 196 | CTT ≥ TTT | L ≥ F | 0.80 | |
| 1919 | 640 | GCT ≥ GTT | A ≥ V | 0.80 | |
| ndhG | 166 | 56 | CAT ≥ TAT | H ≥ Y | 0.80 |
| 320 | 107 | ACA ≥ ATA | T ≥ I | 0.80 | |
| petL | 119 | 40 | CCT ≥ CTT | P ≥ L | 0.86 |
| psbF | 77 | 26 | TCT ≥ TTT | S ≥ F | 1.00 |
| rpl20 | 308 | 103 | TCA ≥ TTA | S ≥ L | 0.86 |
| rpoA | 830 | 277 | TCA ≥ TTA | S ≥ L | 1.00 |
| rpoB | 338 | 113 | TCT ≥ TTT | S ≥ F | 1.00 |
| 551 | 184 | TCA ≥ TTA | S ≥ L | 1.00 | |
| 566 | 189 | TCG ≥ TTG | S ≥ L | 1.00 | |
| 2426 | 809 | TCA ≥ TTA | S ≥ L | 0.86 | |
| rpoC1 | 41 | 14 | TCA ≥ TTA | S ≥ L | 1.00 |
| rpoC2 | 1681 | 561 | CAT ≥ TAT | H ≥ Y | 0.86 |
| 2030 | 677 | ACT ≥ ATT | T ≥ I | 1.00 | |
| 2314 | 772 | CGG ≥ TGG | R ≥ W | 1.00 | |
| 4183 | 1395 | CTT ≥ TTT | L ≥ F | 0.80 | |
| rps2 | 248 | 83 | TCA ≥ TTA | S ≥ L | 1.00 |
| rps16 | 209 | 70 | TCA ≥ TTA | S ≥ L | 0.83 |
| Gene | A.A Position | Citrus aurantiifolia | Rhus chinensis | Dodonaea viscosa | Boswellia Sacra | Leitneria floridana | Azadirachta indica | Ailanthus altissima |
|---|---|---|---|---|---|---|---|---|
| Codon (A.A) Conversion | ||||||||
| atpF | 31 | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) |
| clpP | 187 | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) |
| MatK | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | |
| ndhA | 358 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) |
| ndhB | 50 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) |
| 156 | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | CCA (P) ≥ CTA (L) | |
| 196 | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | |
| 249 | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | |
| 419 | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | |
| ndhD | 1 | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) | ACG (T) ≥ ATG (M) |
| 128 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | |
| ndhG | 107 | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) | ACA (T) ≥ ATA (I) |
| rpoA | 278 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) |
| rpoB | 113 | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) | TCT (S) ≥ TTT (F) |
| 184 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | |
| 809 | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | TCG (S) ≥ TTG (L) | |
| rpoC1 | 14 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) |
| rpoC2 | 563 | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) | CAT (H) ≥ TAT (Y) |
| rps14 | 27 | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) | TCA (S) ≥ TTA (L) |
| Number | Size | Position 1 | Type | Position 2 | Location 1 (2) | Region |
|---|---|---|---|---|---|---|
| 1 | 48 | 95,957 | F | 95,975 | ycf2 | IRa |
| 2 | 48 | 153,174 | F | 153,192 | ycf2 | IRb |
| 3 | 37 | 103,326 | F | 125,821 | rps12/trnV-GAC(ndhA*) | IRa/SSC |
| 4 | 30 | 95,957 | F | 95,993 | ycf2 | IRa |
| 5 | 30 | 153,174 | F | 153,210 | ycf2 | IRb |
| 6 | 29 | 50,944 | F | 50,972 | trnL-UAA* | LSC |
| 7 | 29 | 58,040 | F | 58,078 | rbcL | LSC |
| 8 | 28 | 115,434 | F | 115,460 | ycf1 | SSC |
| 9 | 26 | 39,399 | F | 39,625 | psbZ/trnG-UCC | LSC |
| 10 | 25 | 71,153 | F | 71,178 | trnP-GGG/psaJ | LSC |
| 11 | 23 | 47,036 | F | 103,323 | ycf3**(rps12/trnV-GAC) | LSC/IRa |
| 12 | 23 | 112,456 | F | 112,488 | rrn4.5/rrn5 | IRa |
| 13 | 23 | 136,686 | F | 136,718 | rrn5/rrn4.5 | IRb |
| 14 | 22 | 11,749 | F | 11,771 | trnR-UCU/atpA | LSC |
| 15 | 21 | 248 | F | 270 | trnH-GUG/psbA | LSC |
| 16 | 21 | 9541 | F | 38,293 | trnS-GCU (trnS-UGA) | LSC |
| 17 | 21 | 41,956 | F | 44,180 | psaB(psaA) | LSC |
| 18 | 21 | 49,678 | F | 49,699 | trnL-UAA* | LSC |
| 19 | 20 | 1945 | F | 1965 | trnK-UUU | LSC |
| 20 | 20 | 15,166 | F | 92,503 | atpH/atpI(ycf2) | LSC |
| 21 | 20 | 47,039 | F | 125,821 | ycf3**(rps15) | LSC/IRa |
| 22 | 20 | 88,907 | F | 160,270 | rpl2 | IRa/IRb |
| 25 | 48 | 31,790 | P | 31,790 | petN/psbM | LSC |
| 26 | 48 | 95,957 | P | 153,174 | ycf2 | IRa/IRb |
| 27 | 48 | 95,975 | P | 153,192 | ycf2 | IRa/IRb |
| 28 | 37 | 125,821 | P | 145,834 | ndhA*(trnV-GAC/rps12) | SSC/IRb |
| 29 | 36 | 30,970 | P | 30,970 | petN/psbM | LSC |
| 30 | 30 | 72,117 | P | 72,117 | rpl33/rps18 | LSC |
| 31 | 30 | 95,957 | P | 153,174 | ycf2 | IRa/IRb |
| 32 | 30 | 95,993 | P | 153,210 | ycf2 | IRa/IRb |
| 33 | 27 | 542 | P | 571 | trnH-GUG/psbA | LSC |
| 34 | 25 | 11,403 | P | 11,430 | trnS-GCU/trnR-UCU | LSC |
| 35 | 24 | 4867 | P | 4897 | trnK-UUU/rps16 | LSC |
| 36 | 24 | 9535 | P | 48,164 | trnS-GCU(psaA/ycf3) | LSC |
| 37 | 23 | 47,036 | P | 145,851 | ycf3**(trnV-GAC/rps12) | LSC/IRb |
| 38 | 23 | 51,804 | P | 119,066 | trnF-GAA/ndhJ(rpl32/trnL-UAG) | LSC/SSC |
| 39 | 23 | 112,456 | P | 136,686 | rrn4.5/rrn5 | IRa/IRb |
| 40 | 23 | 112,488 | P | 136,718 | rrn4.5/rrn5 | IRa/IRb |
| 41 | 22 | 39,195 | P | 39,195 | psbZ/trnG-UCC | LSC |
| 42 | 20 | 15,166 | P | 156,674 | atpH(ycf2) | LSC/IRb |
| 43 | 20 | 38,361 | P | 48,100 | trnS-UGA(trnS-GGA) | LSC |
| 44 | 20 | 88,907 | P | 88,907 | rpl2 | IRa |
| 45 | 20 | 107,097 | P | 107,130 | rrn16/trnI-GAU | IRa |
| 46 | 23 | 39,184 | R | 39,184 | psbZ/trnG-UCC | LSC |
| 47 | 21 | 9751 | R | 9751 | trnS-GCU/trnR-UCU | LSC |
| 48 | 21 | 51,281 | R | 51,281 | trnL-UAA/trnF-GAA | LSC |
| 49 | 21 | 85,055 | R | 85,055 | rps8/rpl14 | LSC |
| 50 | 20 | 53,712 | R | 53,712 | ndhC | LSC |
| 51 | 20 | 9385 | R | 13,356 | psbI(atpA/atpF) | LSC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saina, J.K.; Li, Z.-Z.; Gichira, A.W.; Liao, Y.-Y. The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree. Int. J. Mol. Sci. 2018, 19, 929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19040929
Saina JK, Li Z-Z, Gichira AW, Liao Y-Y. The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree. International Journal of Molecular Sciences. 2018; 19(4):929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19040929
Chicago/Turabian StyleSaina, Josphat K., Zhi-Zhong Li, Andrew W. Gichira, and Yi-Ying Liao. 2018. "The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree" International Journal of Molecular Sciences 19, no. 4: 929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19040929