Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Stress Pathways and Malignant Plasma Cell Fitness
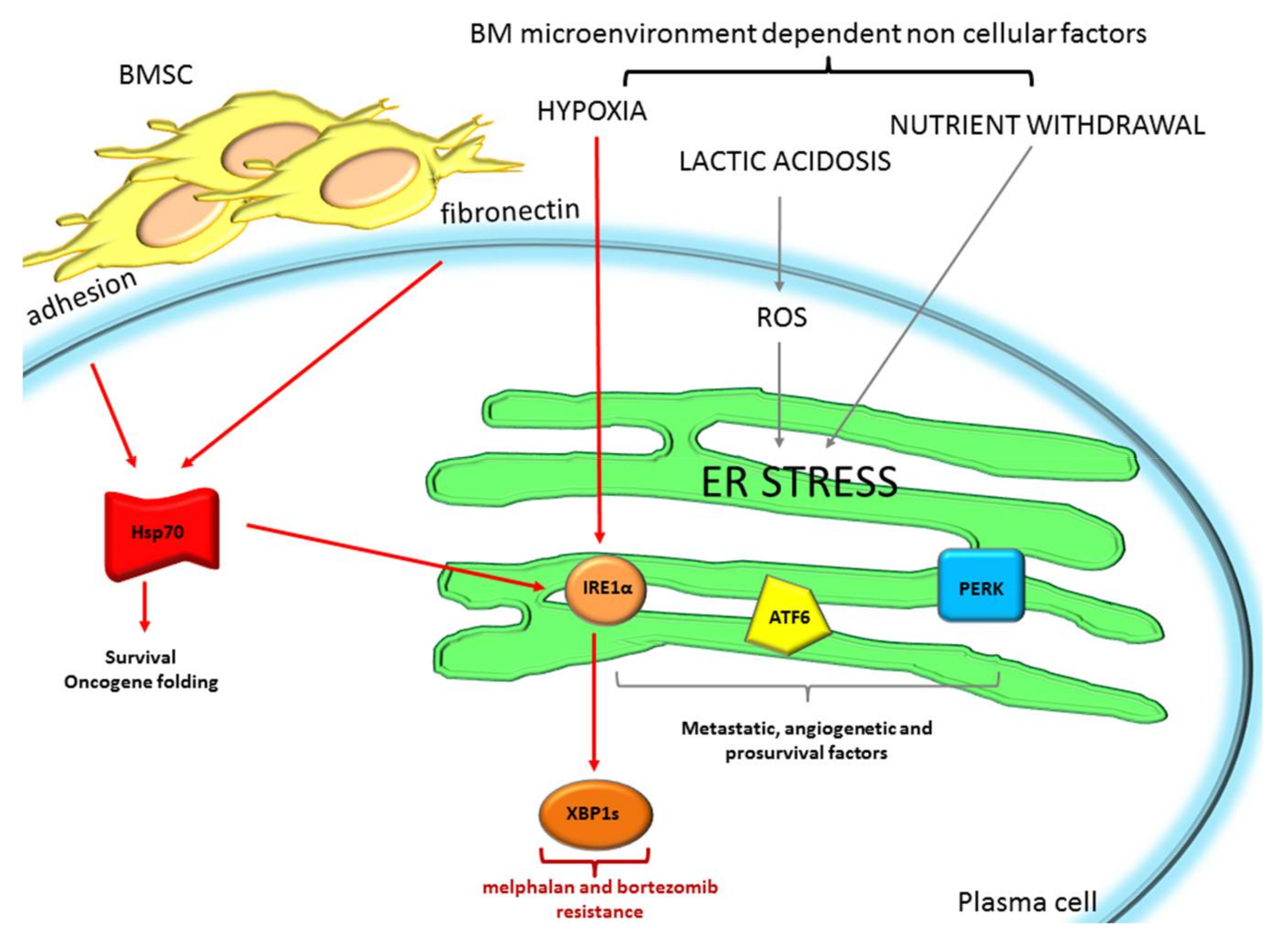
2.1. The Endoplasmic Reticulum Stress/Unfolded Protein Response: A Life Vest for MM Cells to Escape from Chemotherapeutic Agents
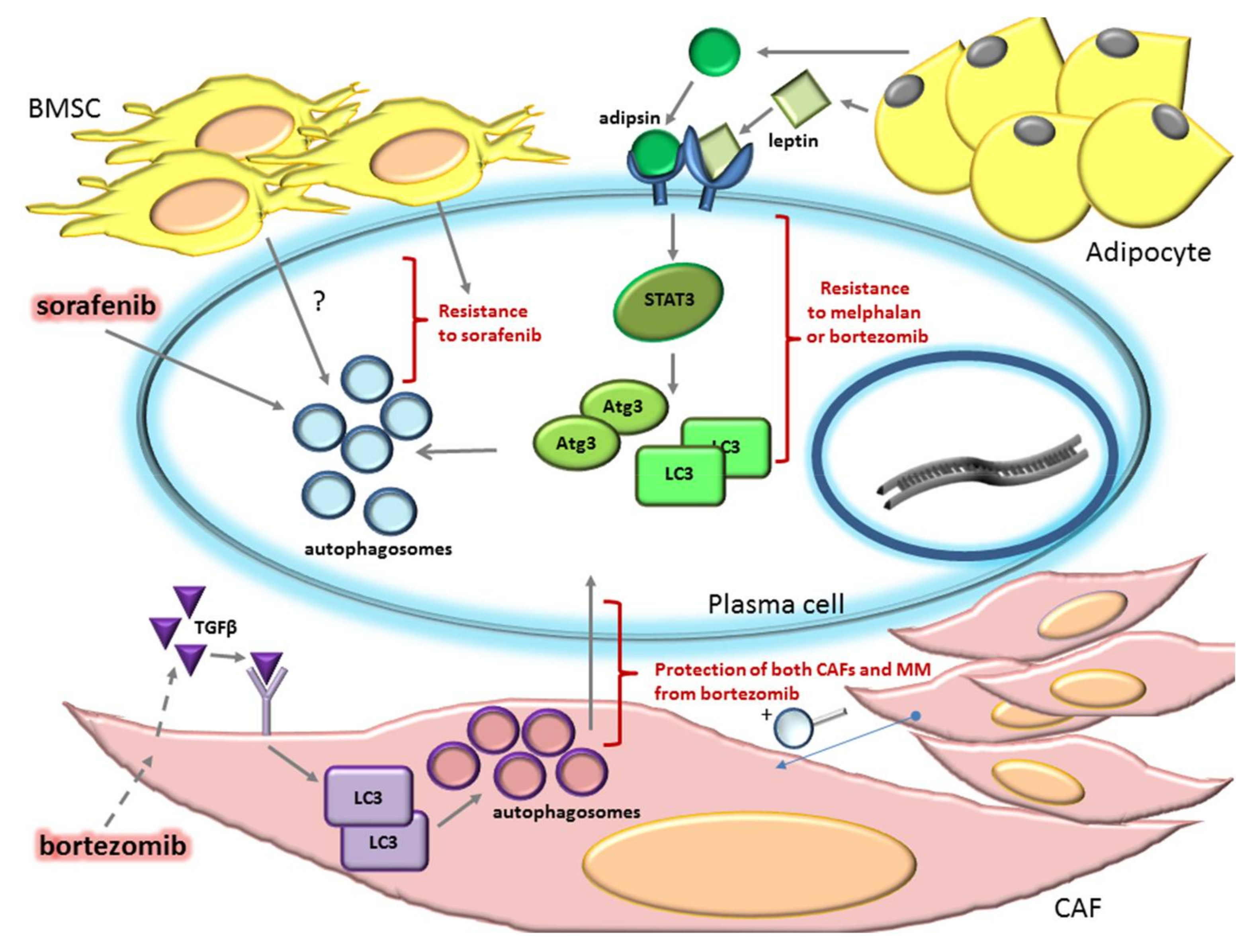
2.2. Autophagy Can Sustain Drug Resistance in MM
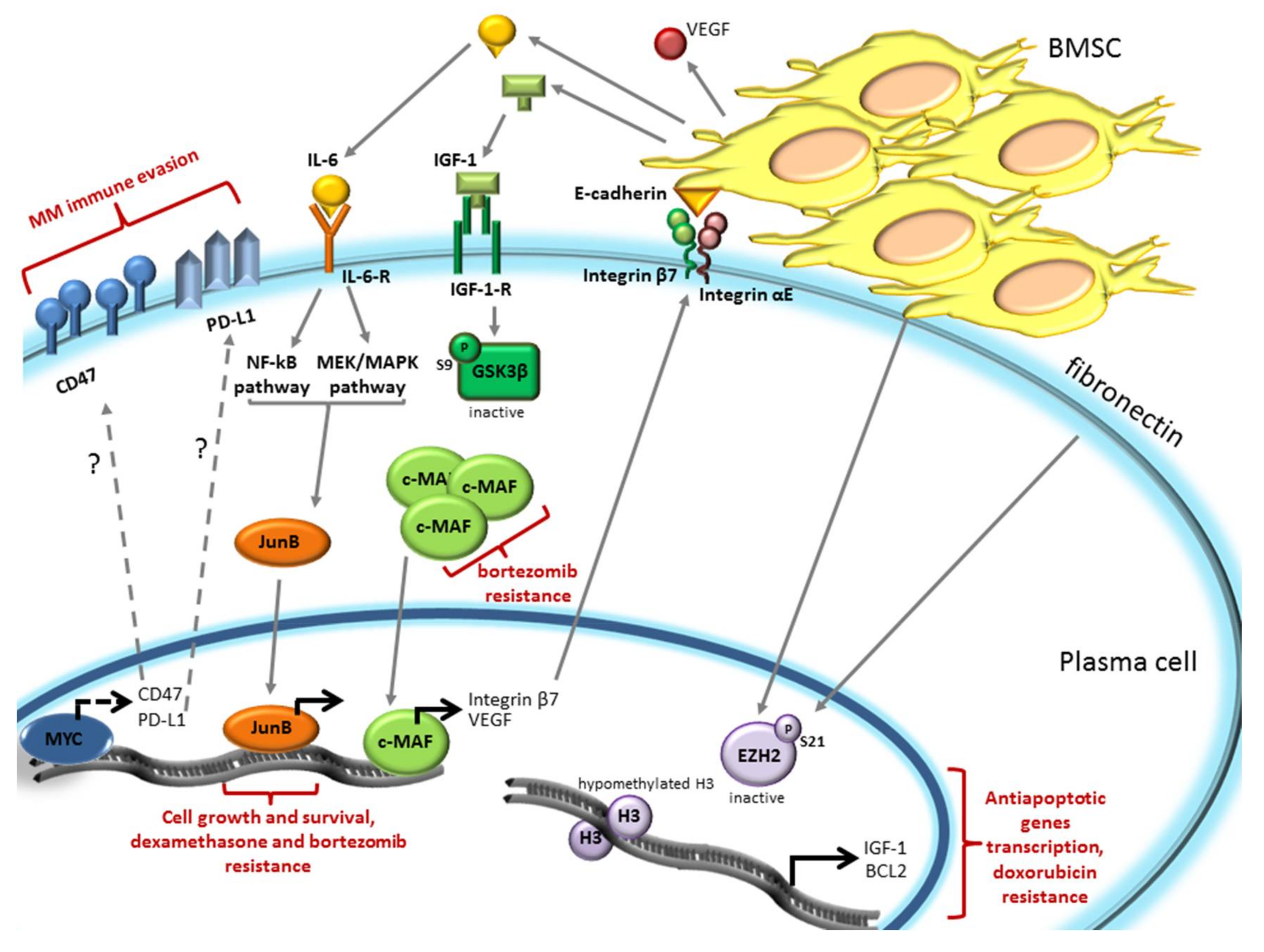
3. The Bone Marrow Microenvironment Dependent Modulation of Gene Expression Induces Drug Resistance in Plasma Cells
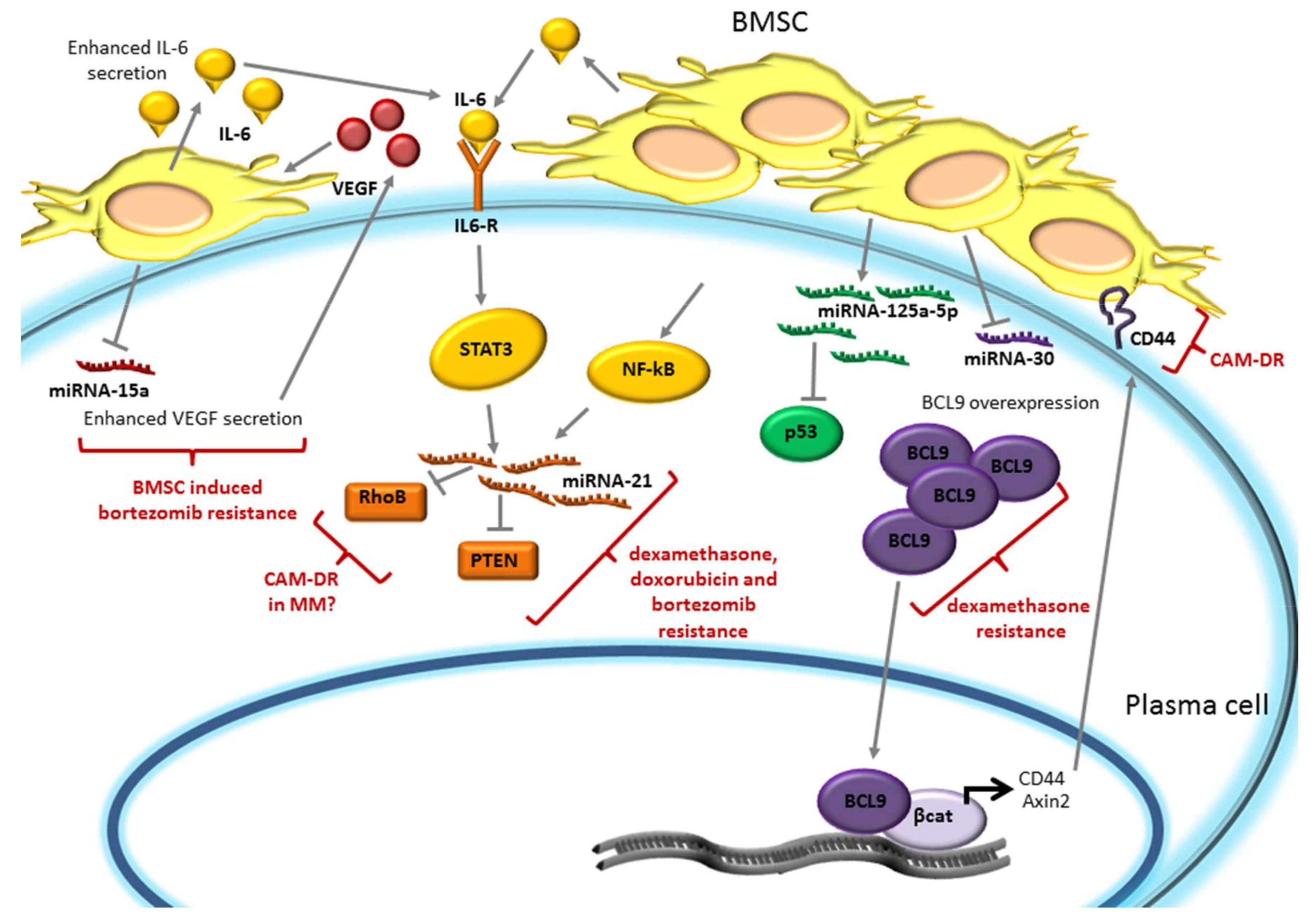
4. miRNA Expression is Modulated by the Bone Marrow Microenvironment and Promotes Plasma Cell Drug Resistance
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| 4-OHT | 4-hydroxytamoxifen |
| ATF4 | transcription factor 4 |
| ATF6 | Activating Transcription Factor-6 |
| Atg3 | Atrogin 3 |
| Atg5 | Atrogin 5 |
| Atg7 | Atrogin 7 |
| BAX | Bcl2 Associated X |
| Bcl2 | B-cell lymphoma 2 |
| BCl9 | B Cell CLL/Lymphoma 9 |
| Bcl-XL | B-cell lymphoma-extra large |
| BM | Bone Marrow |
| BMP6 | Bone Morphogenetic Protein 6 |
| BMSC | Bone Marrow Stromal Cells |
| CAFs | Cancer Associated Fibroblasts |
| CAM-DR | cell adhesion-mediated drug resistance |
| CD47 | Cluster of Differentiation 47 |
| CDKN1A/p21 | cyclin dependent kinase inhibitor 1A |
| CHOP | C/EBP-homologous protein |
| c-MAF | Avian Muscoloaponeurotic Fibrosarcoma |
| CQ | Chloroquine |
| eIF2α | eukaryotic translation initiation factor 2α |
| ER | Endoplasmic Reticulum |
| ERAD | ER-associated degradation |
| EZH2 | Enhancer of Zeste Homolog 2 |
| FAM46C | Family with Sequence Similarity 46 Member C |
| FGF2 | Fibroblast Growth Factor 2 |
| FOXO3 | Forkhead Box O3 |
| GADD54A | Growth Arrest and DNA-Damage Inducible 45 Alpha |
| GC | Glucocorticoid |
| Grp78/Bip | Bglucose regulated protein 78/Binding immunoglobulin protein |
| GSK3β | Glycogen synthase kinase 3β |
| H3K27me3 | Histone H3 trimethylated in Lysine 27 |
| HCC | Hepatocellular carcinoma |
| HIF1α | Hypoxia-inducible factor 1α |
| HSP70 | Heat Shock Protein 70 |
| HSPs | Heat shock chaperone proteins |
| IGF-1 | Insulin Growth Factor 1 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IRE1α | Inositol Requiring Enzyme 1 |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| Mcl1 | Myeloid cell leukemia 1 |
| MDM2 | Mouse Double Minute 2 |
| MEK/MAPK | Mitogen-activated protein kinase kinase/mitogen-activated protein kinase |
| MGUS | Monoclonal Gammopathy of Undetermined Significance |
| MM | Multiple Myeloma |
| mTOR | mammalian target of rapamycine |
| MYC | V-Myc Avian Myelocytomatosis Viral Oncogene |
| MYC T-ALL | MYC-induced T cell acute lymphoblastic leukemia |
| NF-ĸB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| Nrf2 | nuclear factor-erythroid 2-related factor 2 |
| PD-L1 | Programmed death-ligand 1 |
| PERK | kinase PKR-like ER kinase |
| PIs | Proteasome inhibitors |
| POMP | Proteasome maturation proteins |
| PTEN | Phosphate and tensin homolog |
| RhoB | Ras homolog gene family, member B |
| RIDD | IRE1α-dependent decay process |
| ROS | Reactive Oxygen Species |
| ROS | Reactive Oxigen Species |
| sMM | Smouldering Myeloma |
| Sor | Sorafenib |
| SQSTM1/p62 | Sequestosome-1/p62 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TFs | Transcription factors |
| TGFβ | Tumor Growth Factor-β |
| UPS | Ubiquitin Proteasome System |
| VEGF | Vascular Endothelial Growth Factor |
| XBP1 | X-box Binding Protein 1 |
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.-V.; Sonneveld, P.; et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2016, 127, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wasch, R.; Giaccone, L.; Auner, H.W.; Caers, J.; Gramatzki, M.; van de Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Toscani, D.; Mandato, E.; Brancalion, A.; Quotti Tubi, L.; Macaccaro, P.; Cabrelle, A.; Adami, F.; Zambello, R.; Gurrieri, C.; et al. Bone marrow stromal cell-fueled multiple myeloma growth and osteoclastogenesis are sustained by protein kinase CK2. Leukemia 2014, 28, 2094–2097. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kitadate, A.; Abe, F.; Saitoh, H.; Michishita, Y.; Hatano, Y.; Kawabata, Y.; Kitabayashi, A.; Teshima, K.; Kume, M.; et al. Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4 oncogenic axis in multiple myeloma. Cancer Sci. 2017, 108, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed]
- Gooding, S.; Edwards, C.M. New approaches to targeting the bone marrow microenvironment in multiple myeloma. Curr. Opin. Pharmacol. 2016, 28, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bayles, I.; Milcarek, C. Plasma cell formation, secretion, and persistence: The short and the long of it. Crit. Rev. Immunol. 2014, 34, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Sitia, R. The secretory capacity of a cell depends on the efficiency of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 1–15. [Google Scholar] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Carrino, M.; Piazza, F. Role of protein kinases CK1α and CK2 in multiple myeloma: Regulation of pivotal survival and stress-managing pathways. J. Hematol. Oncol. 2017, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Vincenz, L.; Jager, R.; O’Dwyer, M.; Samali, A. Endoplasmic reticulum stress and the unfolded protein response: Targeting the Achilles heel of multiple myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.L.; Moore, H.E.; Dunlop, A.S.; Sharp, S.Y.; Workman, P.; Morgan, G.J.; Davies, F.E. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007, 110, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Hua, Y.; Liu, B. The role of endoplasmic reticulum stress in maintaining and targeting multiple myeloma: A double-edged sword of adaptation and apoptosis. Front. Genet. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Abdel Malek, M.A.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; Abd El-Azeem, H.G.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098–3110. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.B.; Hill, R. GRP78 Influences Chemoresistance and Prognosis in Cancer. Curr. Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, N.C.; Hideshima, T.; Carrasco, D.; Shammas, M.; Auclair, D.; Davies, F.; Mitsiades, N.; Mitsiades, C.; Kim, R.S.; Li, C.; et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood 2004, 103, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fok, J.H.L.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α-XBP1 signaling through a physical interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef] [PubMed]
- Nimmanapalli, R.; Gerbino, E.; Dalton, W.S.; Gandhi, V.; Alsina, M. HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br. J. Haematol. 2008, 142, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.-T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.N.; Rushworth, S.A.; Bowles, K.M.; MacEwan, D.J. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle 2012, 11, 2248–2252. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Li Volti, G. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef] [PubMed]
- Starheim, K.K.; Holien, T.; Misund, K.; Johansson, I.; Baranowska, K.A.; Sponaas, A.M.; Hella, H.; Buene, G.; Waage, A.; Sundan, A.; et al. Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J. 2016, 6, e446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fu, J.; Chen, P.; Ge, X.; Li, Y.; Kuiatse, I.; Wang, H.; Wang, H.; Zhang, X.; Orlowski, R.Z. The Nuclear Factor (Erythroid-derived 2)-like 2 and Proteasome Maturation Protein Axis Mediate Bortezomib Resistance in Multiple Myeloma. J. Biol. Chem. 2015, 290, 29854–29868. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Di Maggio, S.A.; Cabrelle, A.; et al. Protein kinase CK2 inhibition down modulates the NF-κB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib on multiple myeloma and mantle cell lymphoma cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Tubi, L.Q.; Colpo, A.; Pavan, L.; Cabrelle, A.; Ave, E.; Zaffino, F.; Di Maira, G.; Ruzzene, M.; et al. Protein kinase CK2 protects multiple myeloma cells from ER stress-induced apoptosis and from the cytotoxic effect of HSP90 inhibition through regulation of the unfolded protein response. Clin. Cancer Res. 2012, 18, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Autophagy: An Integral Component of the Mammalian Stress Response. J. Biochem. Pharmacol. Res. 2013, 1, 176–188. [Google Scholar] [PubMed]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef] [PubMed]
- Lamy, L.; Ngo, V.N.; Emre, N.C.T.; Shaffer, A.L., III; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O.; et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viability. Mol. Cancer Ther. 2009, 8, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramirez-Labrada, A.; Martinez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Coluccia, A.M.L.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFRβ and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 112, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; De Raeve, H.; Fristedt, C.; Li, Q.; Gruber, A.; Johnsson, P.; Kokaraki, G.; Panzar, M.; Laane, E.; Osterborg, A.; et al. Sorafenib has potent antitumor activity against multiple myeloma in vitro, ex vivo, and in vivo in the 5T33MM mouse model. Cancer Res. 2012, 72, 5348–5362. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; Ceder, S.; Sanchez, C.; Panaretakis, T. Multitargeted therapies for multiple myeloma. Autophagy 2013, 9, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; He, J.; Liu, H.; Lin, P.; Wan, X.; Navone, N.M.; Tong, Q.; Kwak, L.W.; Orlowski, R.Z.; et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 2015, 6, 34329–34341. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGFβ and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; De Veirman, K.; Desantis, V.; di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I.; et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016, 30, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Affer, M.; Chesi, M.; Chen, W.-D.G.; Keats, J.J.; Demchenko, Y.N.; Roschke, A.V.; Van Wier, S.; Fonseca, R.; Bergsagel, P.L.; Kuehl, W.M. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Martelli, M.L.; Gabrea, A.; Qi, Y.; Brents, L.A.; Roschke, A.; Dewald, G.; Kirsch, I.R.; Bergsagel, P.L.; Kuehl, W.M. Diverse karyotypic abnormalities of the c-MYC locus associated with c-MYC dysregulation and tumor progression in multiple myeloma. Proc. Natl. Acad. Sci. USA 2000, 97, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2017. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.L.; Bataille, R. Intergroupe Francophone du Myélome. Rearrangements of the c-MYC oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Frost, P.; Hoang, B.; Benavides, A.; Gera, J.; Lichtenstein, A. IL-6-induced enhancement of c-Myc translation in multiple myeloma cells: Critical role of cytoplasmic localization of the RNA-binding protein hnRNP A1. J. Biol. Chem. 2011, 286, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Eychene, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L.; Shonukan, O.O.; Martelli, M.L.; Brents, L.A.; Chen, T.; Schrock, E.; Ried, T.; Kuehl, W.M. Frequent dysregulation of the c-MAF proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 1998, 91, 4457–4463. [Google Scholar] [PubMed]
- Fonseca, R.; Barlogie, B.; Bataille, R.; Bastard, C.; Bergsagel, P.L.; Chesi, M.; Davies, F.E.; Drach, J.; Greipp, P.R.; Kirsch, I.R.; et al. Genetics and cytogenetics of multiple myeloma: A workshop report. Cancer Res. 2004, 64, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kienast, J.; Berdel, W.E. c-MAF in multiple myeloma: An oncogene enhancing tumor-stroma interactions. Cancer Cell 2004, 5, 109–110. [Google Scholar] [CrossRef]
- Qiang, Y.-W.; Ye, S.; Chen, Y.; Buros, A.F.; Edmonson, R.; van Rhee, F.; Barlogie, B.; Epstein, J.; Morgan, G.J.; Davies, F.E. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood 2016, 128, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L. Many multiple myelomas: Making more of the molecular mayhem. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Mateos, M.-V.; Gutierrez, N.C.; Rajkumar, S.V.; San Miguel, J.F. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013, 121, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Rocques, N.; Abou Zeid, N.; Sii-Felice, K.; Lecoin, L.; Felder-Schmittbuhl, M.-P.; Eychene, A.; Pouponnot, C. GSK-3-mediated phosphorylation enhances MAF-transforming activity. Mol. Cell 2007, 28, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Herath, N.I.; Rocques, N.; Garancher, A.; Eychene, A.; Pouponnot, C. GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J. 2014, 4, e175. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Investig. 2015, 125, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Zhang, L.; An, G.; Meng, H.; Han, Y.; Xie, Z.; Xu, Y.; Li, C.; Yu, Z.; Chang, H.; et al. Bone marrow stromal cells protect myeloma cells from bortezomib induced apoptosis by suppressing microRNA-15a expression. Leuk. Lymphoma 2011, 52, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Karaa, Z.S.; Iacovoni, J.S.; Bastide, A.; Lacazette, E.; Touriol, C.; Prats, H. The VEGF IRESes are differentially susceptible to translation inhibition by miR-16. RNA 2009, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.-K.A.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. miRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 2006, 1, e116. [Google Scholar] [CrossRef] [PubMed]
- Kulda, V.; Pesta, M.; Topolcan, O.; Liska, V.; Treska, V.; Sutnar, A.; Rupert, K.; Ludvikova, M.; Babuska, V.; Holubec, L., Jr.; et al. Relevance of miR-21 and miR-143 expression in tissue samples of colorectal carcinoma and its liver metastases. Cancer Genet. Cytogenet. 2010, 200, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Agnelli, L.; Mosca, L.; Fabris, S.; Andronache, A.; Todoerti, K.; Ronchetti, D.; Deliliers, G.L.; Neri, A. Integrative high-resolution microarray analysis of human myeloma cell lines reveals deregulated miRNA expression associated with allelic imbalances and gene expression profiles. Genes Chromosomes Cancer 2009, 48, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, C.; Ju, S.; Wang, Y.; Wang, H.; Zhong, R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk. Lymphoma 2011, 52, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Leone, E.; Morelli, E.; Di Martino, M.T.; Amodio, N.; Foresta, U.; Gulla, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Cimbora-Zovko, T.; Fritz, G.; Mikac, N.; Osmak, M. Downregulation of RhoB GTPase confers resistance to cisplatin in human laryngeal carcinoma cells. Cancer Lett. 2010, 295, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Loffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermuller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [PubMed]
- Leotta, M.; Biamonte, L.; Raimondi, L.; Ronchetti, D.; Di Martino, M.T.; Botta, C.; Leone, E.; Pitari, M.R.; Neri, A.; Giordano, A.; et al. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J. Cell Physiol. 2014, 229, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhou, L.; Wang, C.; Xu, X.; Jiang, J.; Qin, Y.; Wang, X.; Zhao, C.; Shao, S. Involvement of miR-125a in resistance to daunorubicin by inhibiting apoptosis in leukemia cell lines. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-J.; Lin, J.; Zhu, D.; Wang, X.; Brooks, D.; Chen, M.; Chu, Z.-B.; Takada, K.; Ciccarelli, B.; Admin, S.; et al. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/β-catenin/BCL9 pathway. Cancer Res. 2014, 74, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manni, S.; Carrino, M.; Semenzato, G.; Piazza, F. Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance. Int. J. Mol. Sci. 2018, 19, 1512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051512
Manni S, Carrino M, Semenzato G, Piazza F. Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance. International Journal of Molecular Sciences. 2018; 19(5):1512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051512
Chicago/Turabian StyleManni, Sabrina, Marilena Carrino, Gianpietro Semenzato, and Francesco Piazza. 2018. "Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance" International Journal of Molecular Sciences 19, no. 5: 1512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051512