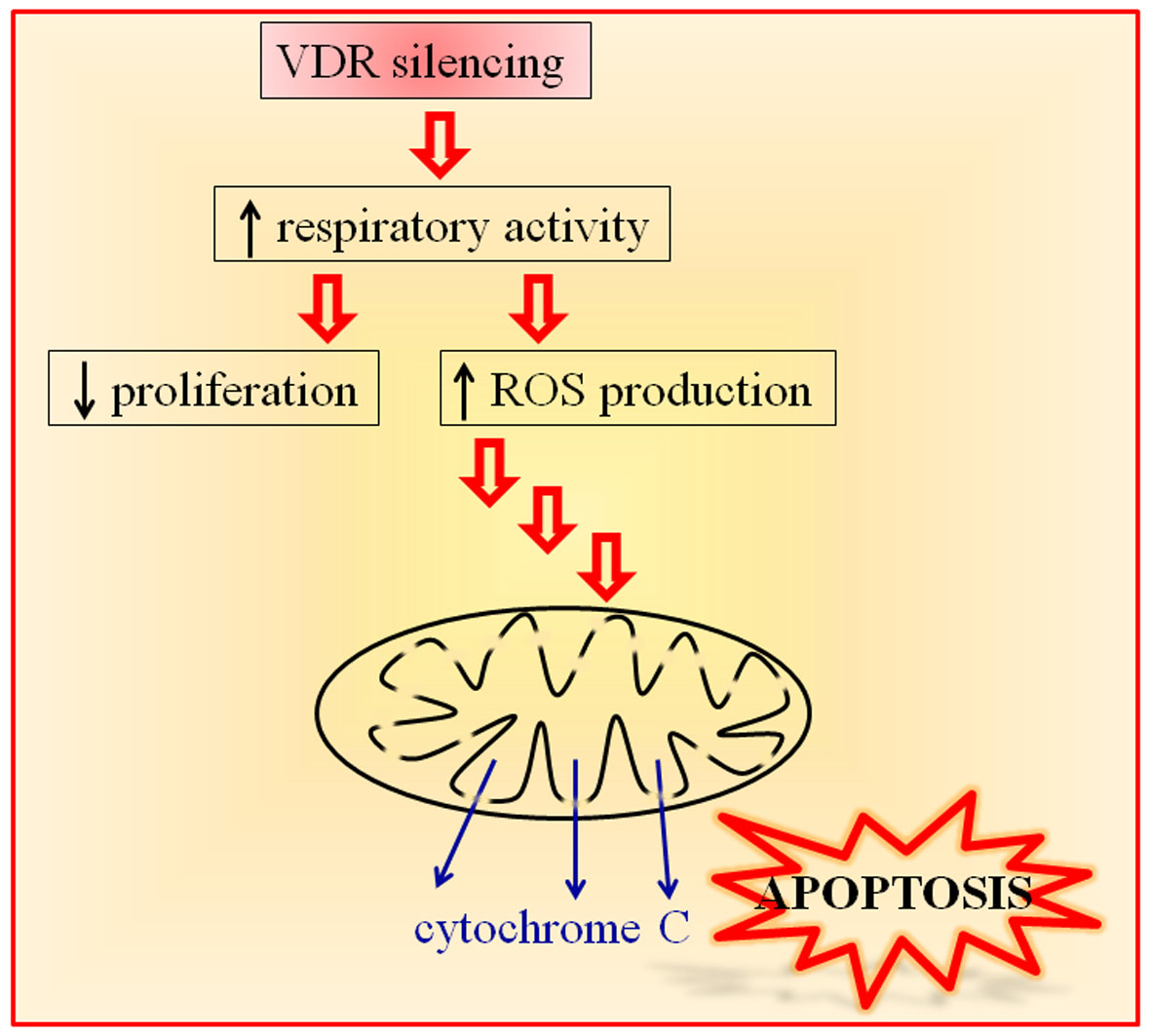
Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
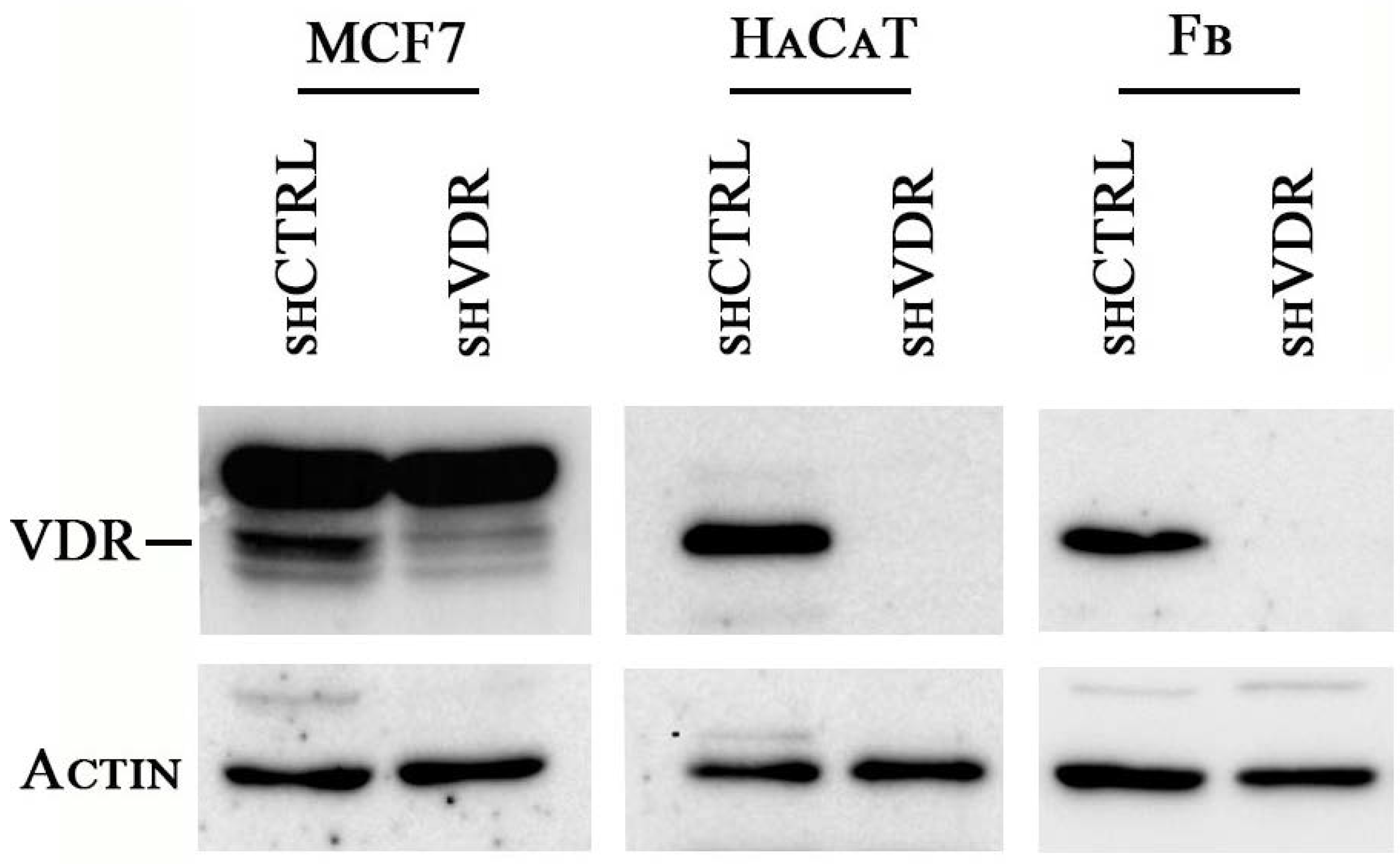
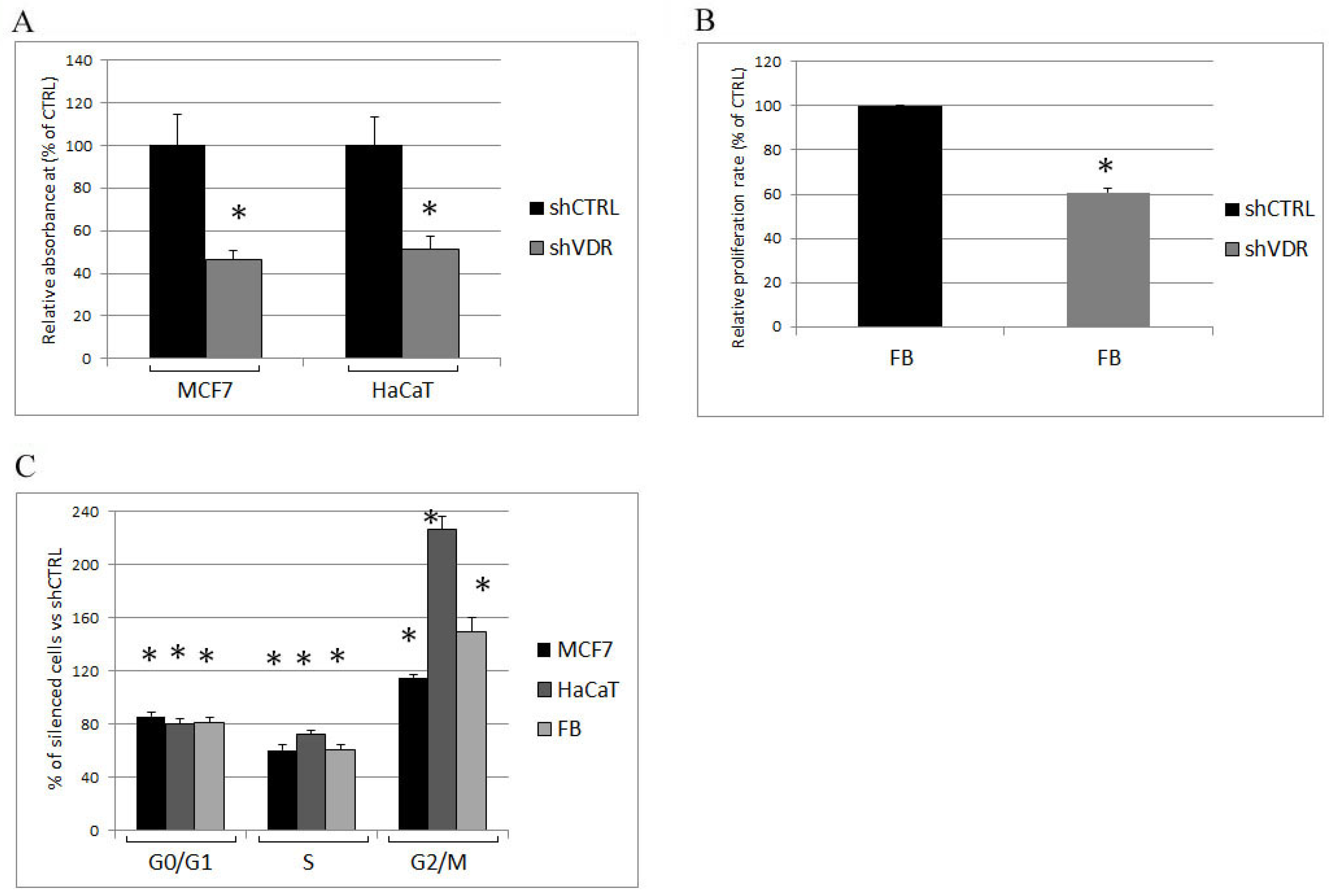
2.1. Two Different Human Cell Lines and Human Primary Cells Silenced for VDR Strongly Reduce Their Proliferation Rate
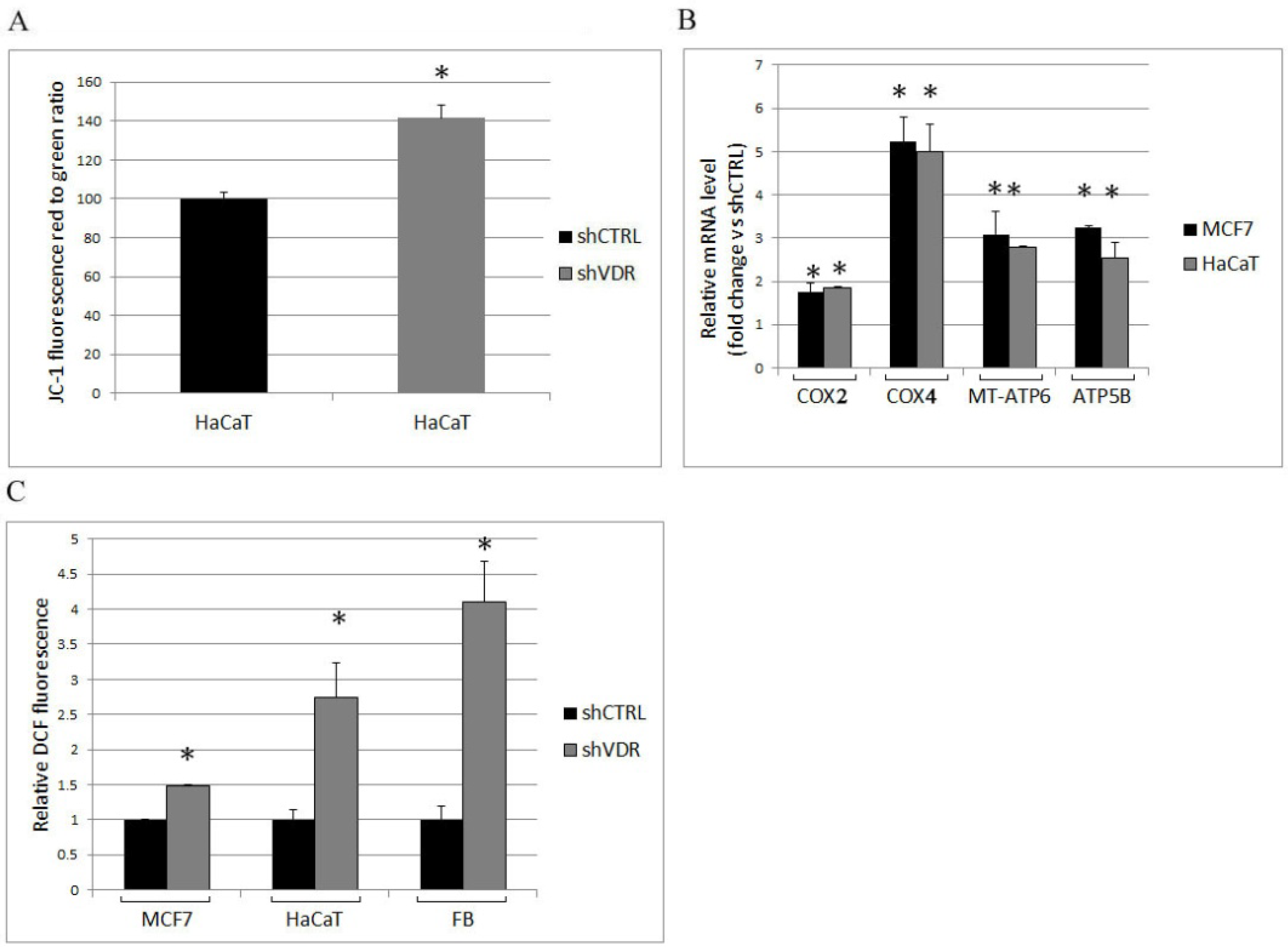
2.2. The Ablation of VDR Enhances Mitochondrial Respiratory Activity and the Production of Reactive Oxygen Species
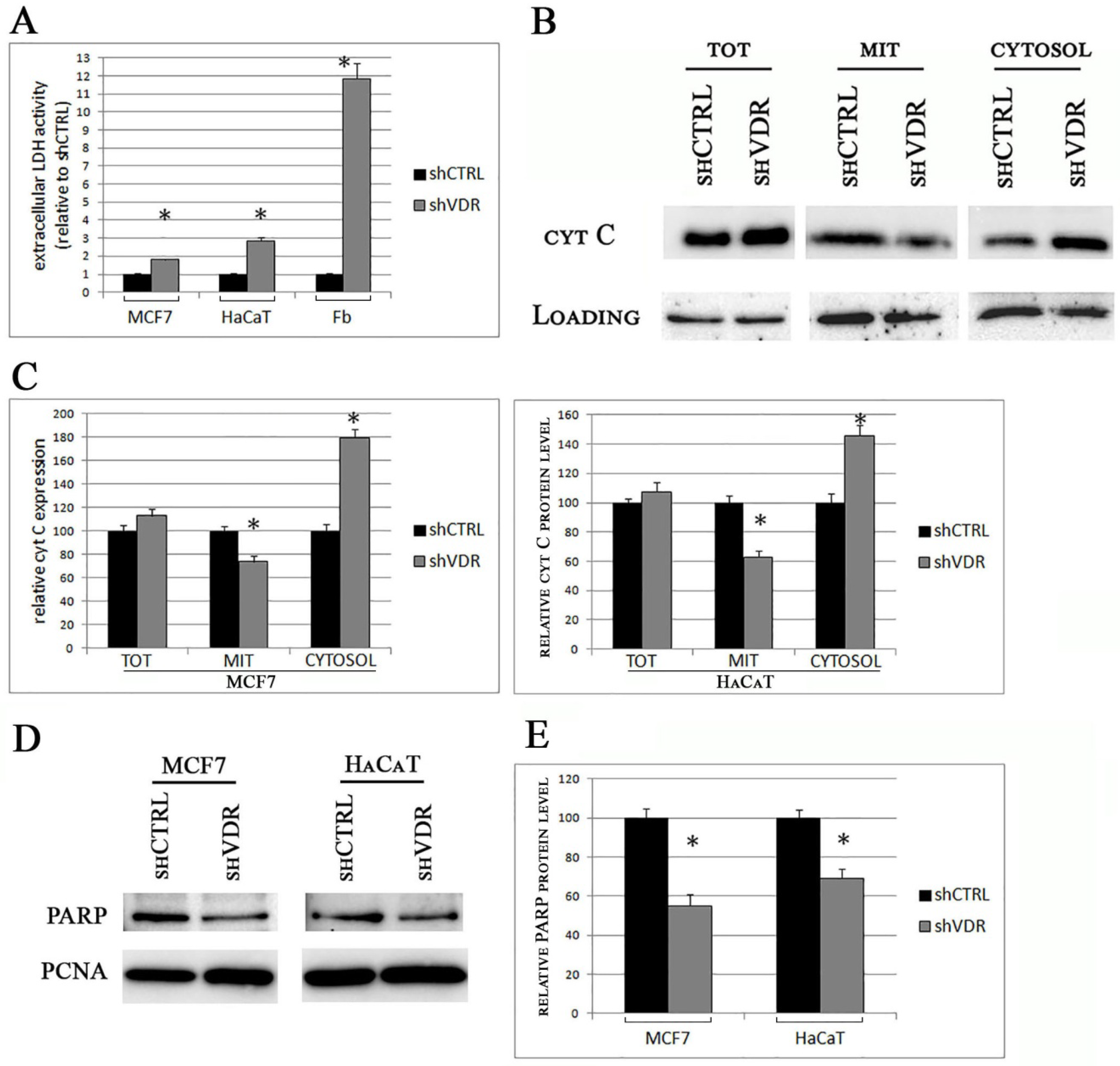
2.3. The Silencing of VDR Triggers Long-Term Cellular Damage and Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentiviral-Mediated shRNA Targeting
4.3. Extract Preparation and Western Blotting Analysis
4.4. Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Proliferation Assay
4.6. Cell Cycle Analysis
4.7. Cytofluorimetric Evaluation of the Mitochondrial Membrane Potential
4.8. Measurement of Intracellular ROS Production
4.9. Toxicity Assay (LDH Release)
4.10. Bands Quantification and Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Barthel, T.K.; Mathern, D.R.; Whitfield, G.K.; Haussler, C.A.; Hopper, H.A.; Hsieh, J.-C.; Slater, S.A.; Hsieh, G.; Kaczmarska, M.; Jurutka, P.W.; et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J. Steroid Biochem. Mol. Biol. 2007, 103, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Jurutka, P.W.; Whitfield, G.K.; Myskowski, S.M.; Eichhorst, K.R.; Dominguez, C.E.; Haussler, C.A.; Haussler, M.R. Liganded VDR induces CYP3A4 in small intestinal and colon cancer cells via DR3 and ER6 vitamin D responsive elements. Biochem. Biophys. Res. Commun. 2002, 299, 730–738. [Google Scholar] [CrossRef]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)2vitamin D3: Genomic and non-genomic mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.; Sitrin, M.D. Vitamin D’s role in cell proliferation and differentiation. Nutr. Rev. 2008, 66, S116–S124. [Google Scholar] [CrossRef] [PubMed]
- Trochoutsou, A.I.; Kloukina, V.; Samitas, K.; Xanthou, G. Vitamin-D in the Immune System: Genomic and Non-Genomic Actions. Mini Rev. Med. Chem. 2015, 15, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Kriebitzsch, C.; Verlinden, L.; Eelen, G.; van Schoor, N.M.; Swart, K.; Lips, P.; Meyer, M.B.; Pike, J.W.; Boonen, S.; Carlberg, C.; et al. 1,25-Dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine β-synthase. J. Bone Miner. Res. 2011, 26, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Bozic, M.; Guzmán, C.; Benet, M.; Sánchez-Campos, S.; García-Monzón, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016, 65, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tayyari, F.; Gowda, G.A.N.; Raftery, D.; McLamore, E.S.; Shi, J.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. 1,25-dihydroxyvitamin D regulation of glucose metabolism in Harvey-ras transformed MCF10A human breast epithelial cells. J. Steroid Biochem. Mol. Biol. 2013, 138, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; De Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial localization of vitamin D receptor in human platelets and differentiated megakaryocytes. PLoS ONE 2010, 5, e8670. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial translocation of vitamin D receptor is mediated by the permeability transition pore in human keratinocyte cell line. PLoS ONE 2013, 8, e54716. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, M.; Destefanis, M.; Morena, D.; Foglizzo, V.; Forneris, M.; Pescarmona, G.; Silvagno, F. The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation. PLoS ONE 2014, 9, e115816. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, M.; Viano, M.; Casarin, S.; Castagnoli, C.; Pescarmona, G.; Silvagno, F. Mitochondrial and lipogenic effects of vitamin D on differentiating and proliferating human keratinocytes. Exp. Dermatol. 2015, 24, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, C.J.; Bae, J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Chen, J.; Zhao, L. 1,25-Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur. J. Nutr. 2015, 54, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Soldani, C.; Lazzè, M.C.; Bottone, M.G.; Tognon, G.; Biggiogera, M.; Pellicciari, C.E.; Scovassi, A.I. Poly(ADP-ribose) polymerase cleavage during apoptosis: When and where? Exp. Cell Res. 2001, 269, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Basit, S. Vitamin D in health and disease: A literature review. Br. J. Biomed. Sci. 2013, 70, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Masuyama, R.; Bouillon, R.; Carmeliet, G. Physiological functions of vitamin D: What we have learned from global and conditional VDR knockout mouse studies. Curr. Opin. Pharmacol. 2015, 22, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Uchida, Y.; Moradian, S.; Crumrine, D.; Elias, P.M.; Bikle, D.D. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J. Investig. Dermatol. 2009, 129, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhu, X.; Shi, Y.; Liu, T.; Chen, Y.; Bhan, I.; Zhao, Q.; Thadhani, R.; Li, Y.C. VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol. Endocrinol. 2013, 27, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Cha, K.M.; Houweling, P.J.; Rao, R.; Mokbel, N.; Lin, M.; Clifton-Bligh, R.J.; Gunton, J.E. Vitamin D Receptor Ablation and Vitamin D Deficiency Result in Reduced Grip Strength, Altered Muscle Fibers, and Increased Myostatin in Mice. Calcif. Tissue Int. 2015, 97, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.E.; Szeto, F.L.; Zhang, W.; Ye, H.; Kong, J.; Zhang, Z.; Sun, X.J.; Li, Y.C. Involvement of the vitamin D receptor in energy metabolism: Regulation of uncoupling proteins. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E820–E828. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, C.J.; Matthews, D.; Broun, E.; Chan, M.; Welsh, J. Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology 2009, 150, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Noolu, B.; Qadri, S.S.Y.H.; Ismail, A. Vitamin D deficiency decreases adiposity in rats and causes altered expression of uncoupling proteins and steroid receptor coactivator3. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt B, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Alimirah, F.; Vaishnav, A.; McCormick, M.; Echchgadda, I.; Chatterjee, B.; Mehta, R.G.; Peng, X. Functionality of unliganded VDR in breast cancer cells: Repressive action on CYP24 basal transcription. Mol. Cell. Biochem. 2010, 342, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Huet, T.; Laverny, G.; Ciesielski, F.; Molnár, F.; Ramamoorthy, T.G.; Belorusova, A.Y.; Antony, P.; Potier, N.; Metzger, D.; Moras, D.; et al. A vitamin D receptor selectively activated by gemini analogs reveals ligand dependent and independent effects. Cell Rep. 2015, 10, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Menegaz, D.; Mizwicki, M.T.; Barrientos-Duran, A.; Chen, N.; Henry, H.L.; Norman, A.W. Vitamin D receptor (VDR) regulation of voltage-gated chloride channels by ligands preferring a VDR-alternative pocket (VDR-AP). Mol. Endocrinol. 2011, 25, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Batie, S.; Lee, J.H.; Jama, R.A.; Browder, D.O.; Montano, L.A.; Huynh, C.C.; Marcus, L.M.; Tsosie, D.G.; Mohammed, Z.; Trang, V.; et al. Synthesis and biological evaluation of halogenated curcumin analogs as potential nuclear receptor selective agonists. Bioorg. Med. Chem. 2013, 21, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Belorusova, A.Y.; Eberhardt, J.; Potier, N.; Stote, R.H.; Dejaegere, A.; Rochel, N. Structural insights into the molecular mechanism of vitamin D receptor activation by lithocholic acid involving a new mode of ligand recognition. J. Med. Chem. 2014, 57, 4710–4719. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. In Mitochondrial Oxidative Phosphorylation; Springer: Berlin/Heidelberg, Germany, 2012; pp. 145–169. [Google Scholar]
- Arnold, S. Cytochrome c oxidase and its role in neurodegeneration and neuroprotection. Adv. Exp. Med. Biol. 2012, 748, 305–339. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Felty, Q.; Narayan, S.; Jayakar, P. Signature of mitochondria of steroidal hormones-dependent normal and cancer cells: Potential molecular targets for cancer therapy. Front. Biosci. 2007, 12, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, A.; Wierzbicka, J.; Ślebioda, T.; Woźniak, M.; Tuckey, R.C.; Slominski, A.T.; Żmijewski, M.A. Vitamin D derivatives enhance cytotoxic effects of H2O2 or cisplatin on human keratinocytes. Steroids 2016, 110, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Shymanskyy, I.O.; Lisakovska, O.O.; Mazanova, A.O.; Labudzynskyi, D.O.; Khomenko, A.V.; Veliky, M.M. Prednisolone and vitamin D3 modulate oxidative metabolism and cell death pathways in blood and bone marrow mononuclear cells. Ukr. Biochem. J. 2016, 88, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Zeeli, T.; Langberg, M.; Rotem, C.; David, M.; Koren, R.; Ravid, A. Vitamin D inhibits captopril-induced cell detachment and apoptosis in keratinocytes. Br. J. Dermatol. 2011, 164, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Uberti, F.; Lattuada, D.; Morsanuto, V.; Nava, U.; Bolis, G.; Vacca, G.; Squarzanti, D.F.; Cisari, C.; Molinari, C. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J. Clin. Endocrinol. Metab. 2014, 99, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.-A.; de Assis, A.M.; Wyse, A.T.S. 1,25-Dihydroxyvitamin D3 prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Nutr. Res. 2017, 38, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Uberti, F.; Bardelli, C.; Morsanuto, V.; Ghirlanda, S.; Molinari, C. Role of vitamin D3 combined to alginates in preventing acid and oxidative injury in cultured gastric epithelial cells. BMC Gastroenterol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Trivedi, T.; Lin, R.C.; Fong-Yee, C.; Nolte, R.; Manibo, J.; Chen, Y.; Hossain, M.; Horas, K.; Dunstan, C.; et al. Loss of the vitamin D receptor in human breast and prostate cancers strongly induces cell apoptosis through downregulation of Wnt/β-catenin signaling. Bone Res. 2017, 5, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon, C.; Rebellato, A.; Urbanet, R.; Guzzardo, V.; Cappellesso, R.; Sasano, H.; Fassina, A.; Fallo, F. Methylation Status of Vitamin D Receptor Gene Promoter in Benign and Malignant Adrenal Tumors. Int. J. Endocrinol. 2015, 2015, 375349. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.; Ayasolla, K.S.; Lan, X.; Sultana-Syed, M.; Chawla, A.; Lederman, R.; Vethantham, V.; Saleem, M.A.; Chander, P.N.; Malhotra, A.; et al. Epigenetic Modulation of Human Podocyte Vitamin D Receptor in HIV Milieu. J. Mol. Biol. 2015, 427, 3201–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurutka, P.W.; Remus, L.S.; Whitfield, G.K.; Thompson, P.D.; Hsieh, J.C.; Zitzer, H.; Tavakkoli, P.; Galligan, M.A.; Dang, H.T.; Haussler, C.A.; et al. The polymorphic N terminus in human vitamin D receptor isoforms influences transcriptional activity by modulating interaction with transcription factor IIB. Mol. Endocrinol. 2000, 14, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Pulito, C.; Terrenato, I.; Di Benedetto, A.; Korita, E.; Goeman, F.; Sacconi, A.; Biagioni, F.; Blandino, G.; Strano, S.; Muti, P.; et al. Cdx2 polymorphism affects the activities of vitamin D receptor in human breast cancer cell lines and human breast carcinomas. PLoS ONE 2015, 10, e0124894. [Google Scholar] [CrossRef] [PubMed]
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhang, Q.; Chen, S.; Fang, B.; Yang, Q.; Chen, C.; Miele, L.; Sarkar, F.H.; Xia, J.; Wang, Z. Mitochondrial dysfunction promotes breast cancer cell migration and invasion through HIF1α accumulation via increased production of reactive oxygen species. PLoS ONE 2013, 8, e69485. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, J.H.; Han, K.-L.; Lee, S.-H.; Hwang, J.-K. Protective effects of panduratin A against oxidative damage of tert-butylhydroperoxide in human HepG2 cells. Biol. Pharm. Bull. 2005, 28, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Destefanis, M.; Viano, M.; Leo, C.; Gervino, G.; Ponzetto, A.; Silvagno, F. Extremely low frequency electromagnetic fields affect proliferation and mitochondrial activity of human cancer cell lines. Int. J. Radiat. Biol. 2015, 91, 964–972. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061672
Ricca C, Aillon A, Bergandi L, Alotto D, Castagnoli C, Silvagno F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. International Journal of Molecular Sciences. 2018; 19(6):1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061672
Chicago/Turabian StyleRicca, Chiara, Alessia Aillon, Loredana Bergandi, Daniela Alotto, Carlotta Castagnoli, and Francesca Silvagno. 2018. "Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health" International Journal of Molecular Sciences 19, no. 6: 1672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061672