Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers

Abstract
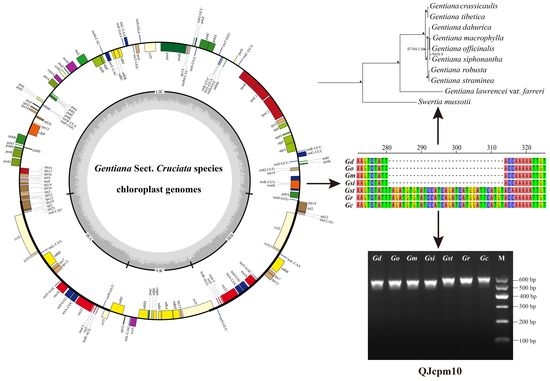
:
1. Introduction
2. Results
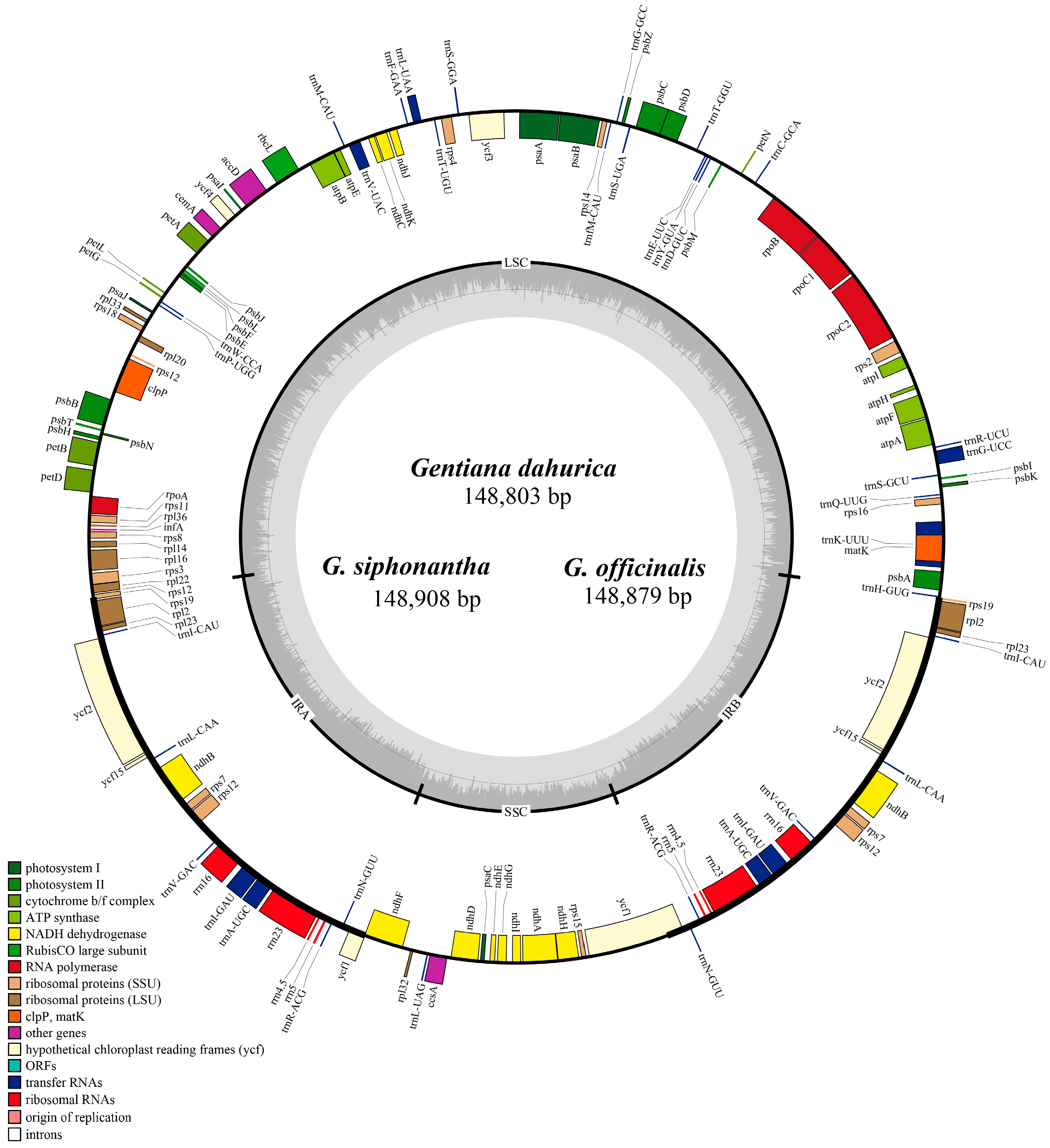
2.1. Complete Chloroplast Genome Features of Sect. Cruciata
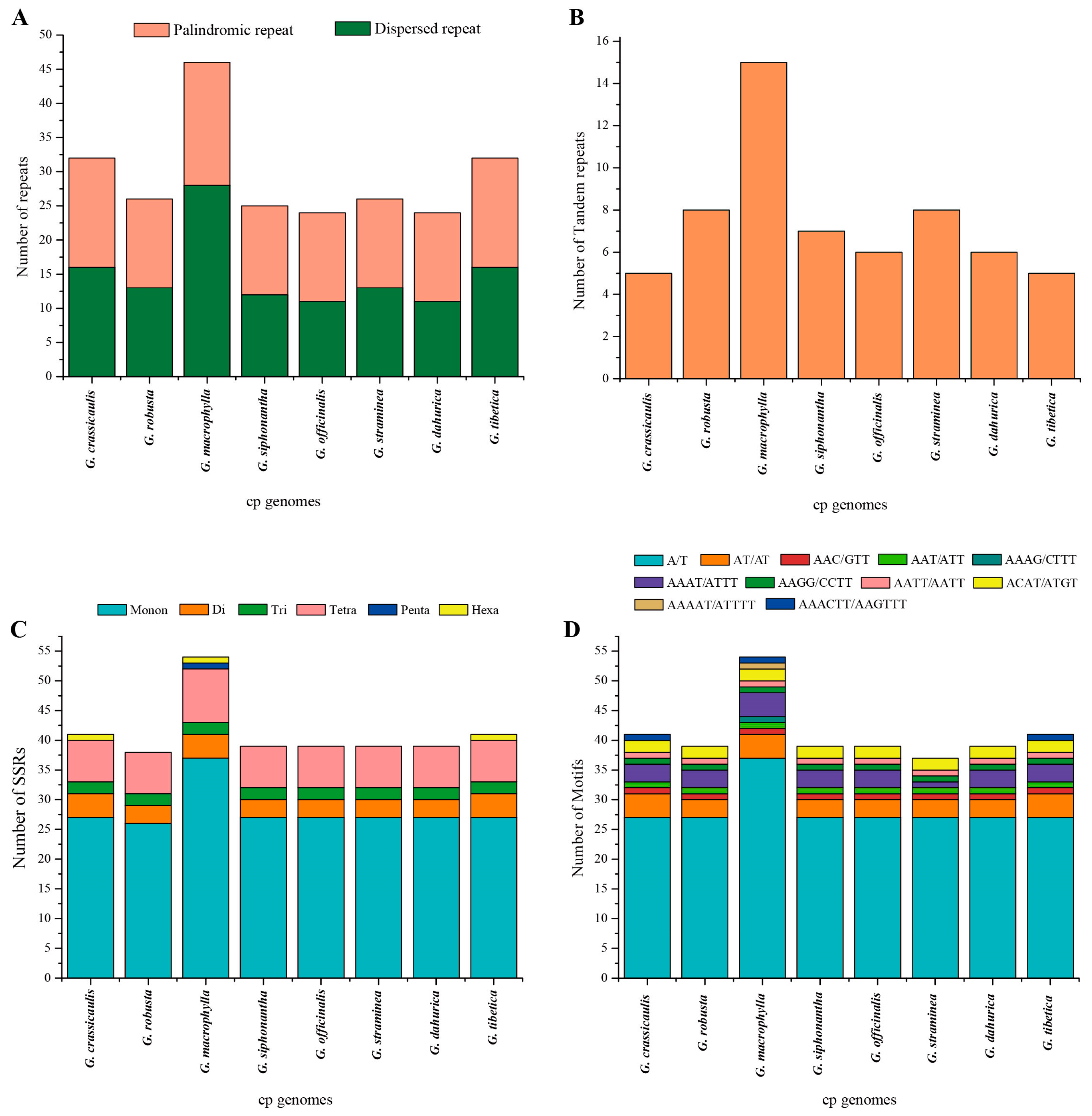
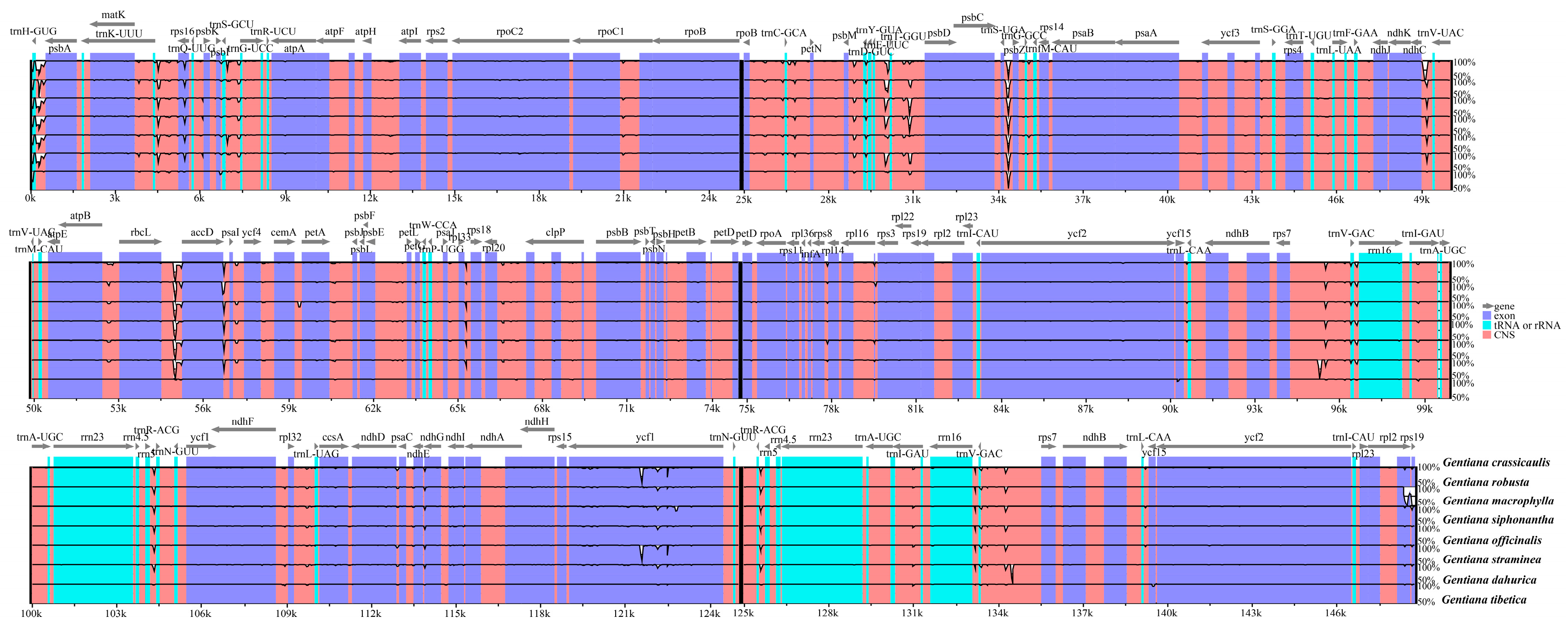
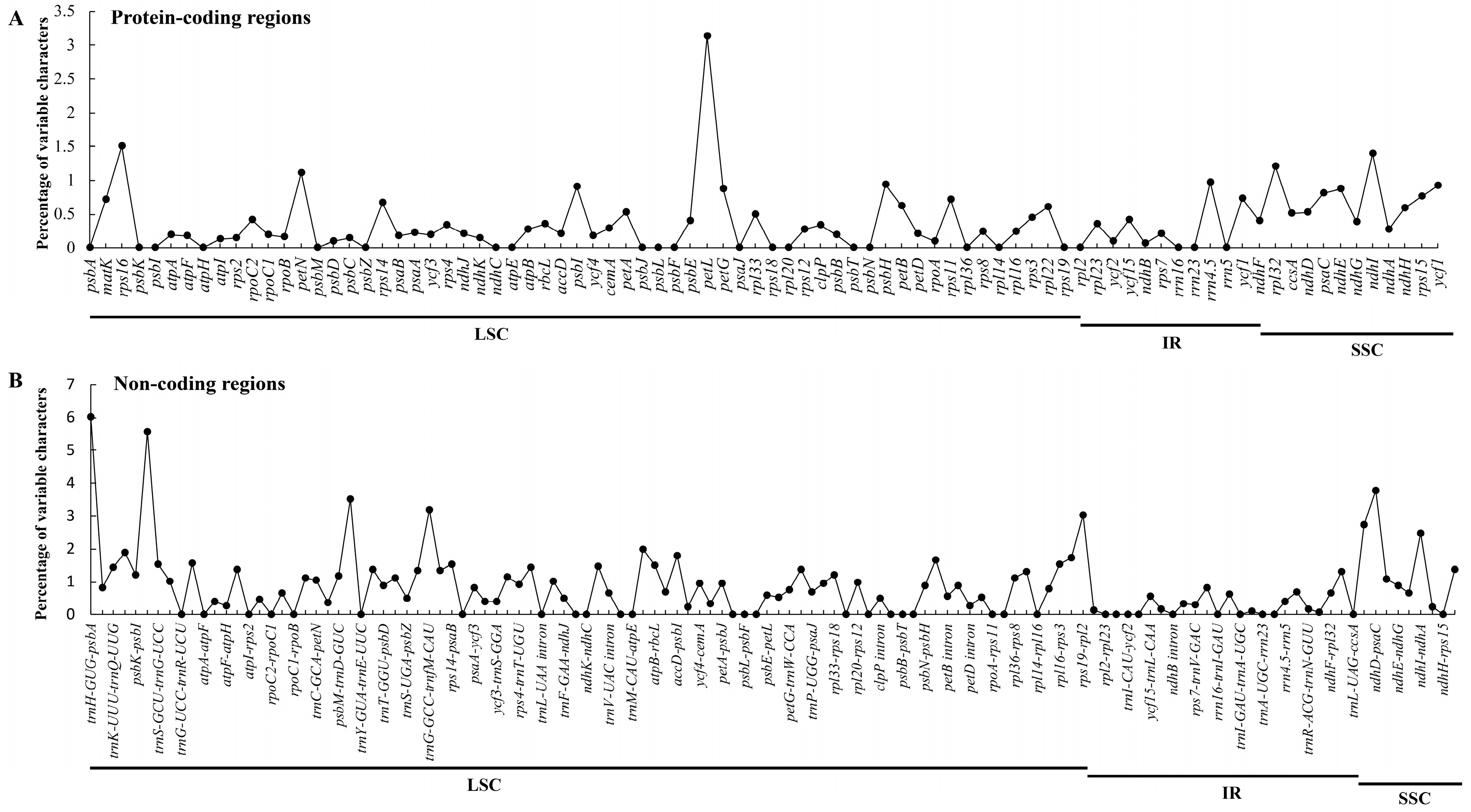
2.2. Comparative Analyses of the Chloroplast Genomes of Species of Sect. Cruciata
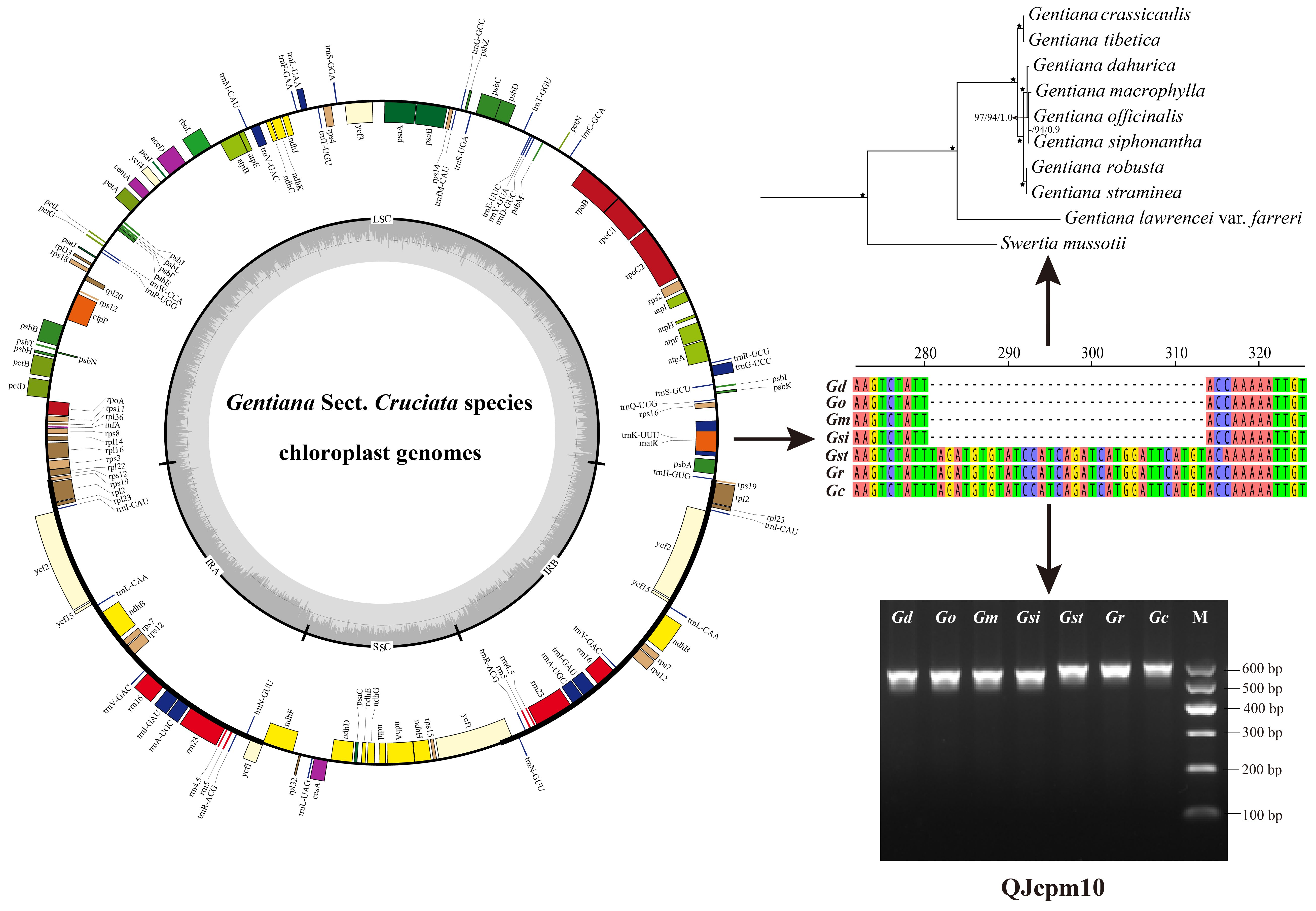
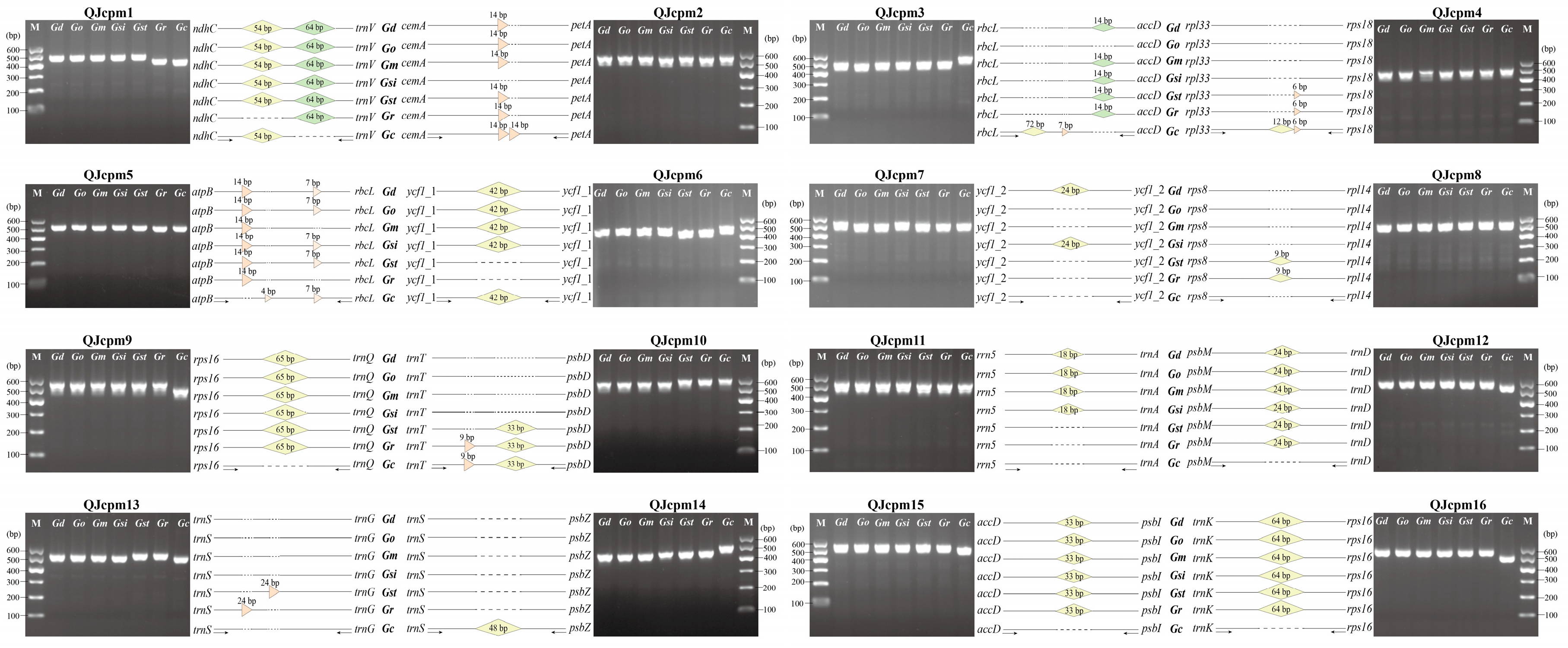
2.3. Development of InDel Markers to Discriminate Species of Sect. Cruciata
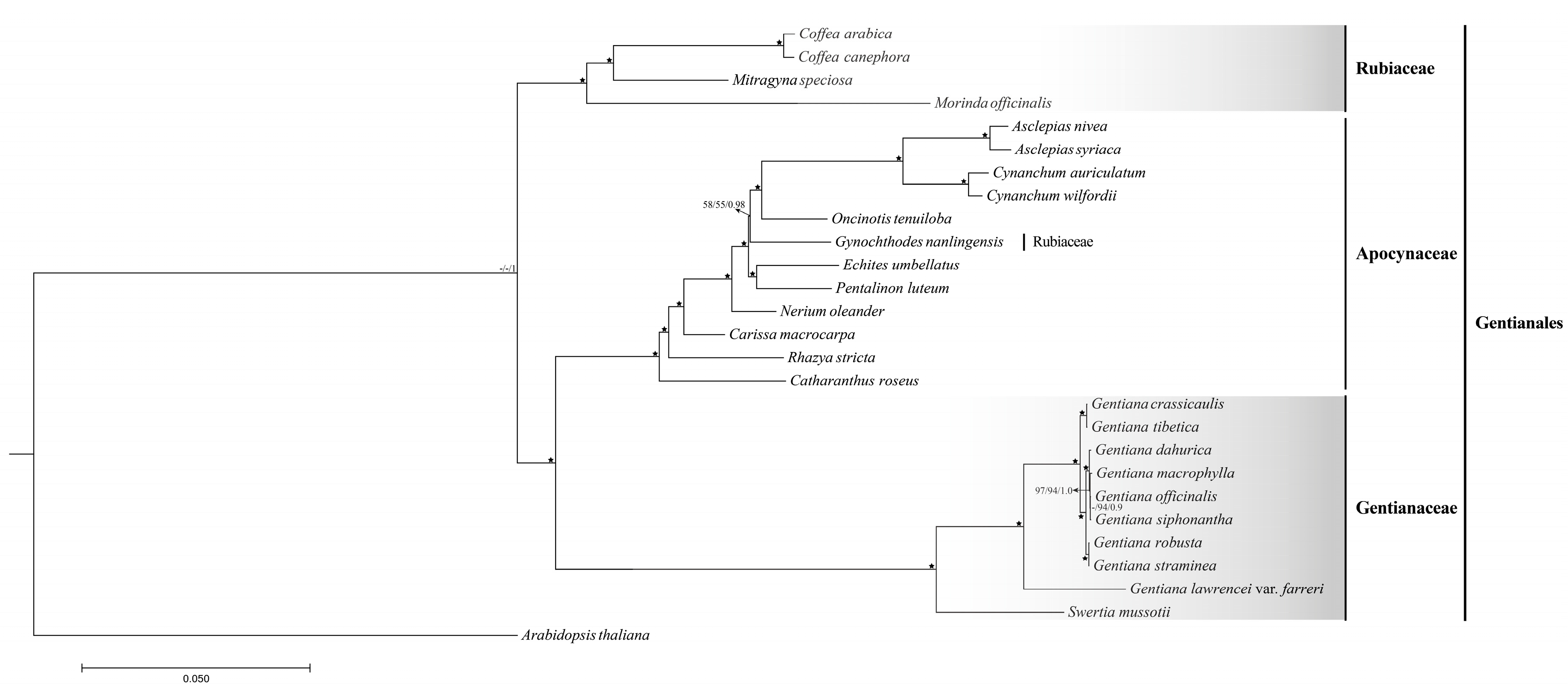
2.4. Phylogenetic Relationships of Species Belong to Sect. Cruciata
3. Discussion
4. Materials and Methods
4.1. Plant Materials and DNA Isolation
4.2. Chloroplast Genome Sequencing, Assembly and Annotation
4.3. Repeat Structure, Genome Comparison and Sequence Divergence
4.4. Development and Validation of the InDel Molecular Marker
4.5. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zhang, X.L.; Wang, Y.J.; Ge, X.J.; Yuan, Y.M.; Yang, H.L.; Liu, J.Q. Molecular phylogeny and biogeography of Gentiana sect. Cruciata (Gentianaceae) based on four chloroplast DNA datasets. Taxon 2009, 58, 862–870. [Google Scholar]
- Ho, T.N.; Liu, S.W. A Worldwide Monograph of Gentiana; Science Press: Beijing, China, 2001. [Google Scholar]
- Ho, T.N.; Pringle, S.J. “Gentianaceae,” Flora of China; Science Press: Beijing, China, 1995; Volume 16, pp. 1–140. [Google Scholar]
- State Pharmacopoeia Commission of the PRC. Pharmacopoeia of P.R. China, Part 1; Chemical Industry Publishing House: Beijing, China, 2015; pp. 270–271. [Google Scholar]
- Hua, W.; Zheng, P.; He, Y.; Cui, L.; Kong, W.; Wang, Z. An insight into the genes involved in secoiridoid biosynthesis in Gentiana macrophylla by RNA-seq. Mol. Biol. Rep. 2014, 41, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Chang-Liao, W.-L.; Chien, C.-F.; Lin, L.-C.; Tsai, T.-H. Isolation of gentiopicroside from Gentianae Radix and its pharmacokinetics on liver ischemia/reperfusion rats. J. Ethnopharmacol. 2012, 141, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhao, Q.; Sun, F.-M.; An, T. Gentiopicrin-producing endophytic fungus isolated from Gentiana macrophylla. Phytomedicine 2009, 16, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, F.; Li, R.; Wang, R. Inhibitory effects of the Gentiana macrophylla (Gentianaceae) extract on rheumatoid arthritis of rats. J. Ethnopharmacol. 2004, 95, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Yang, H.; Liu, J. Confirmation of natural hybrids between Gentiana straminea and G. siphonantha (Gentianaceae) based on molecular evidence. Front. Biol. China 2008, 3, 470–476. [Google Scholar] [CrossRef]
- Hu, Q.; Peng, H.; Bi, H.; Lu, Z.; Wan, D.; Wang, Q.; Mao, K. Genetic homogenization of the nuclear ITS loci across two morphologically distinct gentians in their overlapping distributions in the Qinghai-Tibet Plateau. Sci. Rep. 2016, 6, 34244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Su, J.; Wang, Z. Pharmacognostical studies on root of Gentiana siphonantha. Chin. Tradit. Herbal Drugs 2006, 37, 1875–1878. [Google Scholar]
- Nguyen, V.B.; Park, H.-S.; Lee, S.-C.; Lee, J.; Park, J.Y.; Yang, T.-J. Authentication markers for five major Panax species developed via comparative analysis of complete chloroplast genome sequences. J. Agric. Food Chem. 2017, 65, 6298–6306. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.J. Circular chloroplast chromosomes: The grand illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Koo, H.J.; Lee, J.; Lee, S.-C.; Lee, D.Y.; Giang, V.N.L.; Kim, M.; Shim, H.; Park, J.Y.; Yoo, K.-O.; et al. Authentication of Zanthoxylum species based on integrated analysis of complete chloroplast genome sequences and metabolite profiles. J. Agric. Food Chem. 2017, 65, 10350–10359. [Google Scholar] [CrossRef] [PubMed]
- Eguiluz, M.; Rodrigues, N.F.; Guzman, F.; Yuyama, P.; Margis, R. The chloroplast genome sequence from Eugenia uniflora, a Myrtaceae from Neotropics. Plant Syst. Evol. 2017, 303, 1199–1212. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L.; Fazekas, A.J.; et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [Green Version]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Systematic Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, W.; Xie, X.; Lu, Y.; Liu, Y.; Jin, X.; Suo, Z. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 2017, 8, 1148. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Gruenheit, N.; Ahmadinejad, N.; Timmis, J.; Martin, W. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 2005, 138, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, H.-F.; Ge, X.-J. The use of DNA barcoding on recently diverged species in the genus Gentiana (Gentianaceae) in China. PLoS ONE 2016, 11, e0153008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, Q.; Li, F.; Li, Y. DNA molecular identification of botanical origin in Chinese herb Qingjiao. J. Anhui Agric. Sci. 2011, 39, 14609–14612. [Google Scholar]
- Ni, L.; Zhao, Z.; Xu, H.; Chen, S.; Dorje, G. Chloroplast genome structures in Gentiana (Gentianaceae), based on three medicinal alpine plants used in Tibetan herbal medicine. Curr. Genet. 2017, 63, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhao, Z.; Xu, H.; Chen, S.; Dorje, G. The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the Sino-Himalayan subregion. Gene 2016, 577, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, N.; Su, J.; Zhang, H.; Cao, X. The complete chloroplast genome of Gentiana macrophylla. Mitochondrial DNA B 2017, 2, 395–396. [Google Scholar] [CrossRef]
- Xiang, B.; Li, X.; Qian, J.; Wang, L.; Ma, L.; Tian, X.; Wang, Y. The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 2016, 21, 1029. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.-C.; Zhang, Y.-Z.; Geng, H.-M.; Chen, S.-L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-S.; Fu, P.-C.; Zhou, X.-J.; Cheng, Y.-W.; Zhang, F.-Q.; Chen, S.-L.; Gao, Q.-B. The complete plastome sequences of seven species in Gentiana sect. Kudoa (Gentianaceae): Insights into plastid gene loss and molecular evolution. Front. Plant Sci. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia Species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-B.; Li, D.-Z.; Li, H.-T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Moore, M.J.; Zhang, S.; Soltis, P.S.; Soltis, D.E.; Zhao, T.; Meng, A.; Li, X.; Li, J.; Wang, H. Phylogenomic and structural analyses of 18 complete plastomes across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol. Phylogenet. Evol. 2016, 96, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, X.; Hao, Z.; Yang, L.; Zhang, J.; Peng, Y.; Xu, H.; Lu, Y.; Zhang, J.; Shi, J.; et al. Phylogenetic studies and comparative chloroplast genome analyses elucidate the basal position of halophyte Nitraria sibirica (Nitrariaceae) in the Sapindales. Mitochondrial DNA A 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Tsudzuki, T.; Takahashi, S.; Shimada, H.; Kadowaki, K. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: A comparative analysis of four monocot chloroplast genomes. DNA Res. 2004, 11, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Rousseau-Gueutin, M.; Bellot, S.; Martin, G.E.; Boutte, J.; Chelaifa, H.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; Salmon, A.; Ainouche, K. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Mol. Phylogenet. Evol. 2015, 93, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-H.; Liu, Q.; Hu, W.; Wang, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genomes in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Yasui, Y.; Ohnishi, O. Intraspecific cpDNA variations of diploid and tetraploid perennial buckwheat, Fagopyrum cymosum (Polygonaceae). Am. J. Bot. 2003, 90, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-S.; Yun, B.-K.; Yoon, Y.-H.; Hong, S.-Y.; Mekapogu, M.; Kim, K.-H.; Yang, T.-J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.D.O.; Vieira, L.D.N.; Stefenon, V.M.; Oliveira Pedrosa, F.D.; Souza, E.M.D.; Guerra, M.P.; Nodari, R.O. Phylogenomic relationship of feijoa (Acca sellowiana (O.Berg) Burret) with other Myrtaceae based on complete chloroplast genome sequences. Genetica 2017, 145, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Cuenoud, P.; Savolainen, V.; Chatrou, L.W.; Powell, M.; Grayer, R.J.; Chase, M.W. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Am. J. Bot. 2002, 89, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Techen, N.; Parveen, I.; Pan, Z.; Khan, I.A. DNA barcoding of medicinal plant material for identification. Curr. Opin. Biotech. 2014, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Young Park, J.; Sun Lee, Y.; Lee, H.; Park, H.-S.; Jayakodi, M.; Waminal, N.; Hwa Kang, J.; Joo Lee, T.; Sung, S.; et al. Discrimination and authentication of Eclipta prostrata and E. alba based on the complete chloroplast genomes. Plant Breed. Biotech. 2017, 5, 334–343. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, D.; Zhang, X. Pharmacognostical studies on root of Gentiana officinalis. J. Chin. Med. Mater. 2008, 31, 1635–1638. [Google Scholar]
- Xiong, B.; Zhao, Z.; Ni, L.; Gaawe, D.; Mi, M. DNA-based identification of Gentiana robusta and related species. Chin. Med. Mater. 2015, 40, 4680–4685. [Google Scholar]
- Zhang, X.; Ge, X.; Liu, J.; Yuan, Y. Morphological, karyological and molecular delimitation of two gentians: Gentiana crassicaulis versus G. tibetica (Gentianaceae). Acta Phytotaxon. Sin. 2006, 44, 627–640. [Google Scholar] [CrossRef]
- Maria, B.; Bengt, O.; Birgitta, B. Phylogenetic relationships within the Gentianales based on ndhF and rbcL sequences, with particular reference to the Loganiaceae. Am. J. Bot. 2000, 87, 1029–1043. [Google Scholar]
- Yang, L.L.; Li, H.L.; Wei, L.; Yang, T.; Kuang, D.Y.; Li, M.H.; Liao, Y.Y.; Chen, Z.D.; Wu, H.; Zhang, S.Z. A supermatrix approach provides a comprehensive genus-level phylogeny for Gentianales. J. Syst. Evol. 2016, 54, 400–415. [Google Scholar] [CrossRef] [Green Version]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; Depamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. 2), W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Ma, P.-F.; Li, D.-Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, G.; Mao, Y.; Xing, Q.; Cao, M. HomBlocks: A multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics 2018, 110, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. Commands Used in the PAUP Block in PAUP 4.0: Phylogenetic Analysis Using Parsimony 132–135; Smithsonian Institution: Washington, DC, USA, 1998. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]






 in the phylogenetic tree indicated that the support value of branch is 100/100/1.0.
in the phylogenetic tree indicated that the support value of branch is 100/100/1.0.
in the phylogenetic tree indicated that the support value of branch is 100/100/1.0.
in the phylogenetic tree indicated that the support value of branch is 100/100/1.0.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Taxon | G. dahurica | G. siphonantha | G. officinalis | G. straminea |
| Genome length | 148,803 | 148,908 | 148,879 | 148,991 |
| LSC length | 81,154 | 81,121 | 81,119 | 81,240 |
| SSC length | 17,093 | 17,113 | 17,088 | 17,085 |
| IR length | 25,278 | 25,337 | 25,336 | 25,333 |
| Total gene number | 112 | 112 | 112 | 112 |
| No. of protein coding genes | 78 | 78 | 78 | 78 |
| No. of tRNA genes | 30 | 30 | 30 | 30 |
| No. of rRNA genes | 4 | 4 | 4 | 4 |
| GC content in genome (%) | 37.7 | 37.7 | 37.7 | 37.7 |
| Name of Taxon | G. crassicaulis | G. robusta | G. tibetica | G. macrophylla |
| Genome length | 148,776 | 148,911 | 148,765 | 149,916 |
| LSC length | 81,164 | 81,164 | 81,163 | 82,911 |
| SSC length | 17,071 | 17,085 | 17,070 | 17,095 |
| IR length | 25,271 | 25,333 | 25,266 | 24,955 |
| Total gene number | 112 | 112 | 112 | 112 |
| No. of protein coding genes | 78 | 78 | 78 | 78 |
| No. of tRNA genes | 30 | 30 | 30 | 30 |
| No. of rRNA genes | 4 | 4 | 4 | 4 |
| GC content in genome (%) | 37.7 | 37.7 | 37.7 | 37.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Wang, J.; Jia, Y.; Li, W.; Xu, F.; Wang, X. Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers. Int. J. Mol. Sci. 2018, 19, 1962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071962
Zhou T, Wang J, Jia Y, Li W, Xu F, Wang X. Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers. International Journal of Molecular Sciences. 2018; 19(7):1962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071962
Chicago/Turabian StyleZhou, Tao, Jian Wang, Yun Jia, Wenli Li, Fusheng Xu, and Xumei Wang. 2018. "Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers" International Journal of Molecular Sciences 19, no. 7: 1962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071962