Inside the Endometrial Cell Signaling Subway: Mind the Gap(s)

{kind=link}
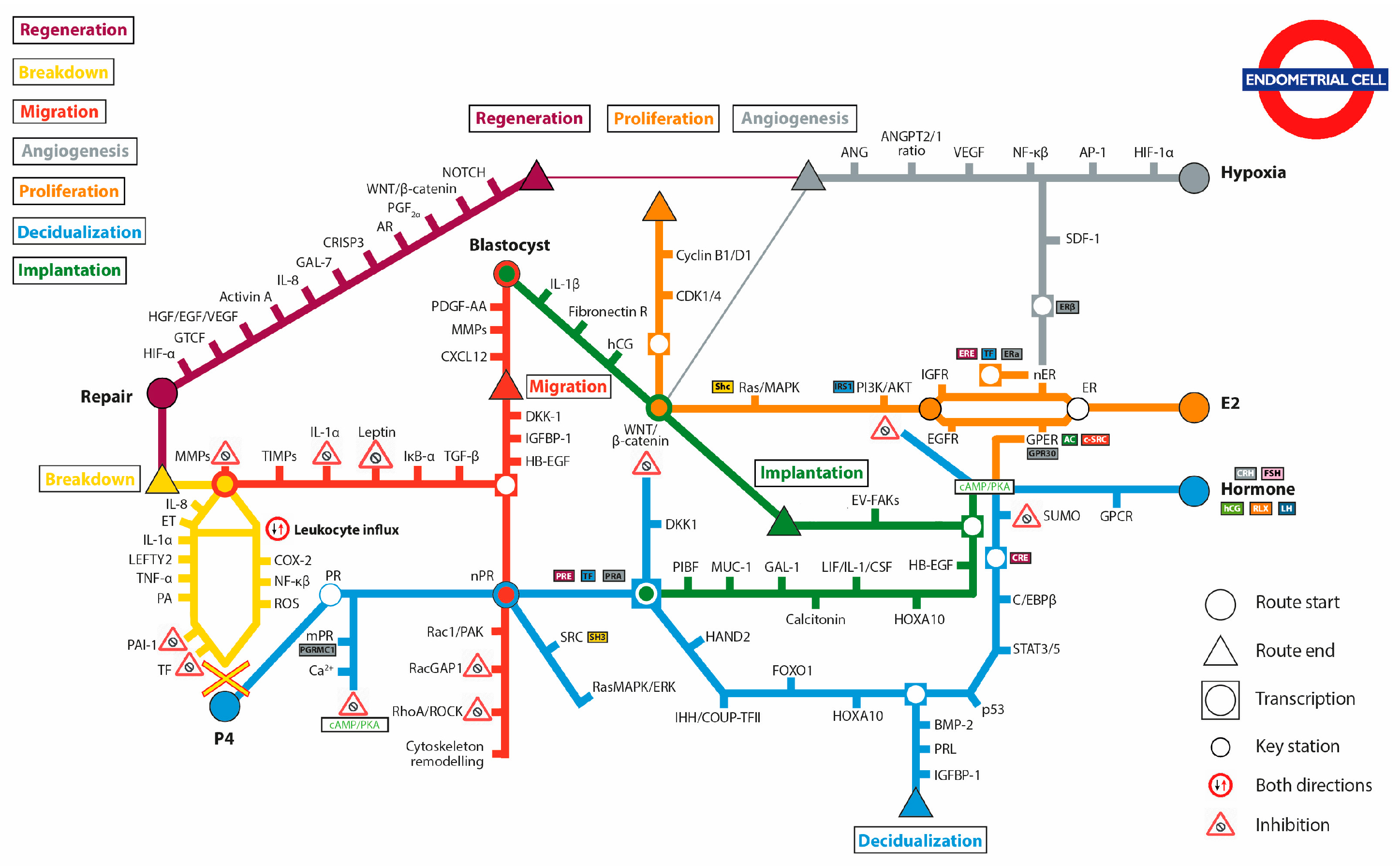
Abstract
:1. Entrance
2. Proliferation Route: Building the Functionalis
3. Decidualization Route: Priming the Endometrium for Implantation
4. Implantation Route: Accepting the Blastocyst
5. Migration Route: Promotion of Blastocyst Invasion
6. Breakdown Route: Shedding the Functionalis
7. Regeneration: Repairing the Functionalis
8. Angiogenesis Route: Building the Endometrial Vascular Network
9. Exit
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, J.; Salamonsen, L.A.; Winship, A.; Menkhorst, E.; Nie, G.; Gargett, C.E.; Dimitriadis, E. Fertile ground: Human endometrial programming and lessons in health and disease. Nat. Rev. Endocrinol. 2016, 12, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Stanley, J.N.; Bhimji, S.S. Anatomy, Pelvis, Uterus. In StatPearls (Internet); StatPearls Publishing LLC: Treasure Island, FL, USA, 2018. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK470297/ (accessed on 4 July 2018).
- Zhu, H.; Hou, C.C.; Luo, L.F.; Hu, Y.J.; Yang, W.X. Endometrial stromal cells and decidualized stromal cells: Origins, transformation and functions. Gene 2014, 551, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bhurke, A.S.; Bagchi, I.C.; Bagchi, M.K. Progesterone-Regulated Endometrial Factors Controlling Implantation. Am. J. Reprod. Immunol. 2016, 75, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashary, N.; Tiwari, A.; Modi, D. Embryo Implantation: War in Times of Love. Endocrinology 2018, 159, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, J.S. A Review of Mechanisms of Implantation. Dev. Reprod. 2017, 21, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maybin, J.A.; Critchley, H.O. Menstrual physiology: Implications for endometrial pathology and beyond. Hum. Reprod. Update 2015, 21, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Berbic, M.; Fraser, I.S. Immunology of normal and abnormal menstruation. Womens Health (Lond.) 2013, 9, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Gargett, C.E.; Nguyen, H.P.; Ye, L. Endometrial regeneration and endometrial stem/progenitor cells. Rev. Endocr. Metab. Disord. 2012, 13, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Reproductive endocrinology: Hypoxia in endometrial repair. Nat. Rev. Endocrinol. 2018, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Young, J.R.; Jaffe, R.B. Strength-duration characteristics of estrogen effects on gonadotropin response to gonadotropin-releasing hormone in women. II. Effects of varying concentrations of estradiol. J. Clin. Endocrinol. Metab. 1976, 42, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Aso, T.; Brenner, P.F.; Cekan, Z.; Landgren, B.M.; Hagenfeldt, K.; Diczfalusy, E. Studies on the pattern of circulating steroids in the normal menstrual cycle. I. Simultaneous assays of progesterone, pregnenolone, dehydroepiandrosterone, testosterone, dihydrotestosterone, androstenedione, oestradiol and oestrone. Acta Endocrinol. (Cph.) 1976, 81, 133–149. [Google Scholar]
- Kurita, T.; Medina, R.; Schabel, A.B.; Young, P.; Gama, P.; Parekh, T.V.; Brody, J.; Cunha, G.R.; Osteen, K.G.; Bruner-Tran, K.L.; et al. The activation function-1 domain of estrogen receptor α in uterine stromal cells is required for mouse but not human uterine epithelial response to estrogen. Differentiation 2005, 73, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Deroo, B.J.; Hansen, K.; Collins, J.; Grissom, S.; Afshari, C.A.; Korach, K.S. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol. Endocrinol. 2003, 17, 2070–2083. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, I.; Jeschke, U.; Shabani, N.; Kuhn, C.; Balle, A.; Kriegel, S.; Kupka, M.S.; Friese, K. Immunohistochemical analysis of estrogen receptor α, estrogen receptor beta and progesterone receptor in normal human endometrium. Acta Histochem. 2004, 106, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Dorostghoal, M.; Ghaffari, H.O.; Marmazi, F.; Keikhah, N. Overexpression of Endometrial Estrogen Receptor-A in The Window of Implantation in Women with Unexplained Infertility. Int. J. Fertil. Steril. 2018, 12, 37–42. [Google Scholar] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Pollard, J.W. Estradiol-17beta regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 15847–15851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Pan, H.; Zhu, L.; Deng, Y.; Pollard, J.W. Progesterone inhibits the estrogen-induced phosphoinositide 3-kinase→AKT→GSK-3beta→cyclin D1→pRB pathway to block uterine epithelial cell proliferation. Mol. Endocrinol. 2005, 19, 1978–1990. [Google Scholar] [CrossRef] [PubMed]
- Winuthayanon, W.; Hewitt, S.C.; Orvis, G.D.; Behringer, R.R.; Korach, K.S. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc. Natl. Acad. Sci. USA 2010, 107, 19272–19277. [Google Scholar] [CrossRef] [PubMed]
- Vrtacnik, P.; Ostanek, B.; Mencej-Bedrac, S.; Marc, J. The many faces of estrogen signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef] [PubMed]
- da Costa e Silva Rde, C.; Moura, K.K.; Ribeiro Junior, C.L.; Guillo, L.A. Estrogen signaling in the proliferative endometrium: Implications in endometriosis. Rev. Assoc. Med. Bras. 2016, 62, 72–77. [Google Scholar] [PubMed]
- Hewitt, S.C.; Li, Y.; Li, L.; Korach, K.S. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor α to estrogen-responsive elements. J. Biol. Chem. 2010, 285, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Groothuis, P.G.; Dassen, H.H.; Romano, A.; Punyadeera, C. Estrogen and the endometrium: Lessons learned from gene expression profiling in rodents and human. Hum. Reprod. Update 2007, 13, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Kashima, H.; Shiozawa, T.; Miyamoto, T.; Suzuki, A.; Uchikawa, J.; Kurai, M.; Konishi, I. Autocrine stimulation of IGF1 in estrogen-induced growth of endometrial carcinoma cells: Involvement of the mitogen-activated protein kinase pathway followed by up-regulation of cyclin D1 and cyclin, E. Endocr. Relat. Cancer 2009, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.G.; DiAugustine, R.P.; Petrusz, P.; Clark, G.C.; Sebastian, J. Estradiol stimulates tyrosine phosphorylation of the insulin-like growth factor-1 receptor and insulin receptor substrate-1 in the uterus. Proc. Natl. Acad. Sci. USA 1996, 93, 12002–12007. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Atta, I.S.; Rowan, B.G.; Desouki, M.M. ERα and ERK1/2 MAP kinase expression in microdissected stromal and epithelial endometrial cells. J. Egypt. Natl. Cancer Inst. 2014, 26, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, S.; Nuedling, S.; van Eickels, M.; Vetter, H.; Meyer, R.; Grohe, C. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J. Biol. Chem. 2000, 275, 18447–18453. [Google Scholar] [CrossRef] [PubMed]
- Yotova, I.Y.; Quan, P.; Leditznig, N.; Beer, U.; Wenzl, R.; Tschugguel, W. Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum. Reprod. 2011, 26, 885–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.X.; Barnes, C.J.; Zhang, Z.; Bao, Y.; Kumar, R.; Santen, R.J. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen–estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, E.; Salvadori, M. Antineoplastic effect of proliferation signal inhibitors: From biology to clinical application. J. Nephrol. 2009, 22, 457–462. [Google Scholar] [PubMed]
- Kim, T.H.; Yu, Y.; Luo, L.; Lydon, J.P.; Jeong, J.W.; Kim, J.J. Activated AKT pathway promotes establishment of endometriosis. Endocrinology 2014, 155, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, H.; Li, M.; Xue, J.; Fu, Y.; Yan, L.; Zhao, X. Normal endometrial stromal cells regulate survival and apoptosis signaling through PI3K/AKt/Survivin pathway in endometrial adenocarcinoma cells in vitro. Gynecol. Oncol. 2011, 123, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol. Cell. Endocrinol. 2016, 429, 10–18. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Y.; Cai, B.; Yang, Y.X.; Liu, X.L.; Wan, X.P. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 2009, 100, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Zucchetti, A.E.; Barosso, I.R.; Boaglio, A.C.; Basiglio, C.L.; Miszczuk, G.; Larocca, M.C.; Ruiz, M.L.; Davio, C.A.; Roma, M.G.; Crocenzi, F.A.; et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ss-D-glucuronide-induced cholestasis. Hepatology 2014, 59, 1016–1029. [Google Scholar] [CrossRef] [PubMed]
- Fabi, F.; Grenier, K.; Parent, S.; Adam, P.; Tardif, L.; Leblanc, V.; Asselin, E. Regulation of the PI3K/Akt pathway during decidualization of endometrial stromal cells. PLoS ONE 2017, 12, e0177387. [Google Scholar] [CrossRef] [PubMed]
- Makker, A.; Goel, M.M.; Nigam, D.; Mahdi, A.A.; Das, V.; Agarwal, A.; Pandey, A.; Gautam, A. Aberrant Akt Activation During Implantation Window in Infertile Women With Intramural Uterine Fibroids. Reprod. Sci. 2018, 25, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Kiewisz, J.; Wasniewski, T.; Kmiec, Z. Participation of WNT and beta-Catenin in Physiological and Pathological Endometrial Changes: Association with Angiogenesis. Biomed. Res. Int. 2015, 2015, 854056. [Google Scholar] [CrossRef] [PubMed]
- Rider, V.; Isuzugawa, K.; Twarog, M.; Jones, S.; Cameron, B.; Imakawa, K.; Fang, J. Progesterone initiates Wnt-beta-catenin signaling but estradiol is required for nuclear activation and synchronous proliferation of rat uterine stromal cells. J. Endocrinol. 2006, 191, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Loose, D.S. Secreted frizzled-related protein 4 regulates two Wnt7a signaling pathways and inhibits proliferation in endometrial cancer cells. Mol. Cancer Res. 2008, 6, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Zhang, L.; Yu, L.; Xie, W.; Man, Y.; Xiong, Y.; Liu, H.; Liu, Y. Estradiol promotes cells invasion by activating beta-catenin signaling pathway in endometriosis. Reproduction 2015, 150, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, P.H.; Wang, Y.; van der Zee, M.; Burger, C.W.; Blok, L.J. Interaction between sex hormones and WNT/beta-catenin signal transduction in endometrial physiology and disease. Mol. Cell. Endocrinol. 2012, 358, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Lehmann, L. Estrogens modulate the gene expression of Wnt-7a in cultured endometrial adenocarcinoma cells. Mol. Nutr. Food Res. 2006, 50, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Munoz, F.J.; Metcalfe, M.J.; Hitschfeld, M.; Olivares, G.; Godoy, J.A.; Inestrosa, N.C. Trolox and 17beta-estradiol protect against amyloid beta-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J. Biol. Chem. 2005, 280, 11615–11625. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, T.; Kajiya, H.; Miyajima, K.; Takei, M.; Tobita, M.; Takekoshi, S.; Osamura, R.Y. The expression of Wnt4 is regulated by estrogen via an estrogen receptor α-dependent pathway in rat pituitary growth hormone-producing cells. Acta Histochem. Cytochem. 2009, 42, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Ashizawa, K.; Fukuhara, T.; Hiroyasu, M.; Tsuzuki, Y.; Tatemoto, H.; Nakada, T.; Nagai, K. Differential expression patterns of Wnt and beta-catenin/TCF target genes in the uterus of immature female rats exposed to 17α-ethynyl estradiol. Toxicol. Sci. 2006, 91, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Hanifi-Moghaddam, P.; Boers-Sijmons, B.; Klaassens, A.H.; van Wijk, F.H.; den Bakker, M.A.; Ott, M.C.; Shipley, G.L.; Verheul, H.A.; Kloosterboer, H.J.; Burger, C.W.; et al. Molecular analysis of human endometrium: Short-term tibolone signaling differs significantly from estrogen and estrogen + progestagen signaling. J. Mol. Med. 2007, 85, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; van der Zee, M.; Fodde, R.; Blok, L.J. Wnt/Beta-catenin and sex hormone signaling in endometrial homeostasis and cancer. Oncotarget 2010, 1, 674–684. [Google Scholar] [PubMed]
- Hou, X.; Tan, Y.; Li, M.; Dey, S.K.; Das, S.K. Canonical Wnt signaling is critical to estrogen-mediated uterine growth. Mol. Endocrinol. 2004, 18, 3035–3049. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Tan, J.; Raja, S.; Halder, J.; Paria, B.C.; Dey, S.K. Estrogen targets genes involved in protein processing, calcium homeostasis, and Wnt signaling in the mouse uterus independent of estrogen receptor-α and -beta. J. Biol. Chem. 2000, 275, 28834–28842. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Hamilton, A.E.; Aghajanova, L.; Vo, K.C.; Nezhat, C.N.; Lessey, B.A.; Giudice, L.C. MicroRNA expression profiling of eutopic secretory endometrium in women with versus without endometriosis. Mol. Hum. Reprod. 2009, 15, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Luo, X.; Toloubeydokhti, T.; Chegini, N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol. Hum. Reprod. 2007, 13, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudanski, P.; Charkiewicz, R.; Tolwinska, A.; Szamatowicz, J.; Charkiewicz, A.; Niklinski, J. Profiling of Selected MicroRNAs in Proliferative Eutopic Endometrium of Women with Ovarian Endometriosis. Biomed. Res. Int. 2015, 2015, 760698. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, M.K.; Mantena, S.R.; Kannan, A.; Bagchi, I.C. Control of uterine cell proliferation and differentiation by C/EBPbeta: Functional implications for establishment of early pregnancy. Cell Cycle 2006, 5, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, P.A.; Critchley, H.O.; Williams, A.R.; Arends, M.J.; Saunders, P.T. New concepts for an old0problem: The diagnosis of endometrial hyperplasia. Hum. Reprod. Update 2017, 23, 232–254. [Google Scholar] [PubMed]
- Plaza-Parrochia, F.; Romero, C.; Valladares, L.; Vega, M. Endometrium and steroids, a pathologic overview. Steroids 2017, 126, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ramathal, C.Y.; Bagchi, I.C.; Taylor, R.N.; Bagchi, M.K. Endometrial decidualization: Of mice and men. Semin. Reprod. Med. 2010, 28, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Hantak, A.M.; Bagchi, I.C.; Bagchi, M.K. Role of uterine stromal-epithelial crosstalk in embryo implantation. Int. J. Dev. Biol. 2014, 58, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brar, A.K.; Frank, G.R.; Kessler, C.A.; Cedars, M.I.; Handwerger, S. Progesterone-dependent decidualization of the human endometrium is mediated by cAMP. Endocrine 1997, 6, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Brosens, J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: A decidualizing affair. J. Endocrinol. 2003, 178, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Yoshimura, Y. Molecular and cellular mechanisms for differentiation and regeneration of the uterine endometrium. Endocr. J. 2008, 55, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Campitiello, M.R.; De Franciscis, P.; Mele, D.; Izzo, G.; Sinisi, A.; Delrio, G.; Colacurci, N. Endometrial LGR7 expression during menstrual cycle. Fertil. Steril. 2011, 95, 2511–2514. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Brosens, J.J. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr. Rev. 2014, 35, 851–905. [Google Scholar] [CrossRef] [PubMed]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, T.; Brosens, J.J.; Ishihara, O. The role of FOXO1 in the decidual transformation of the endometrium and early pregnancy. Med. Mol. Morphol. 2013, 46, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Taylor, R.N.; Bagchi, I.C.; Bagchi, M.K. Regulation of human endometrial stromal proliferation and differentiation by C/EBPbeta involves cyclin E-cdk2 and STAT3. Mol. Endocrinol. 2012, 26, 2016–2030. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.Y.; Brosens, J.J.; Christian, M.; Hills, F.A.; Chamley, L.; Regan, L.; White, J.O. Regulated expression of signal transducer and activator of transcription, Stat5, and its enhancement of PRL expression in human endometrial stromal cells in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2581–2588. [Google Scholar] [CrossRef] [PubMed]
- Pohnke, Y.; Schneider-Merck, T.; Fahnenstich, J.; Kempf, R.; Christian, M.; Milde-Langosch, K.; Brosens, J.J.; Gellersen, B. Wild-type p53 protein is up-regulated upon cyclic adenosine monophosphate-induced differentiation of human endometrial stromal cells. J. Clin. Endocrinol. Metab. 2004, 89, 5233–5244. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Merck, T.; Pohnke, Y.; Kempf, R.; Christian, M.; Brosens, J.J.; Gellersen, B. Physical interaction and mutual transrepression between CCAAT/enhancer-binding protein beta and the p53 tumor suppressor. J. Biol. Chem. 2006, 281, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Sakaguchi, J.; Kiyono, T.; Shimizu, Y.; Maida, Y.; Mizumoto, Y.; Mori, N.; Nakamura, M.; Takakura, M.; Miyake, K.; et al. Forkhead transcription factor FOXO1 is a direct target of progestin to inhibit endometrial epithelial cell growth. Clin. Cancer Res. 2011, 17, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.C.; Fusi, L.; Higham, J.H.; Abdel-Hafiz, H.; Horwitz, K.B.; Lam, E.W.; Brosens, J.J. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc. Natl. Acad. Sci. USA 2006, 103, 16272–16277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Deng, W.B.; Liu, Y.F.; Liang, Y.X.; Fan, Z.M.; Gu, X.W.; Liu, J.L.; Sha, A.G.; Diao, H.L.; Yang, Z.M. Non-coding RNA LINC00473 mediates decidualization of human endometrial stromal cells in response to cAMP signaling. Sci. Rep. 2016, 6, 22744. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Ma, J.; Liu, Y.; Lin, K.; Jiang, X.; Qu, Y.; Lin, J.; Xu, K. Analysis of long non-coding RNA expression profiles using RNA sequencing in ovarian endometriosis. Gene 2018, 673, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Byrne, F.L.; Takenaka, K.; Modesitt, S.C.; Olzomer, E.M.; Mills, J.D.; Farrell, R.; Hoehn, K.L.; Janitz, M. Transcriptome landscape of long intergenic non-coding RNAs in endometrial cancer. Gynecol. Oncol. 2017, 147, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Sigurgeirsson, B.; Amark, H.; Jemt, A.; Ujvari, D.; Westgren, M.; Lundeberg, J.; Gidlof, S. Comprehensive RNA sequencing of healthy human endometrium at two time points of the menstrual cycle. Biol. Reprod. 2017, 96, 24–33. [Google Scholar] [PubMed]
- Prange-Kiel, J.; Rune, G.M.; Zwirner, M.; Wallwiener, D.; Kiesel, L. Regulation of estrogen receptor α and progesterone receptor (isoform A and B) expression in cultured human endometrial cells. Exp. Clin. Endocrinol. Diabetes 2001, 109, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Haluska, G.J.; West, N.B.; Novy, M.J.; Brenner, R.M. Uterine estrogen receptors are increased by RU486 in late pregnant rhesus macaques but not after spontaneous labor. J. Clin. Endocrinol. Metab. 1990, 70, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Mote, P.A.; Balleine, R.L.; McGowan, E.M.; Clarke, C.L. Heterogeneity of progesterone receptors A and B expression in human endometrial glands and stroma. Hum. Reprod. 2000, 15 (Suppl. 3), 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulac-Jericevic, B.; Mullinax, R.A.; DeMayo, F.J.; Lydon, J.P.; Conneely, O.M. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science 2000, 289, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Scott, M.P.; Ribon, V.; Sherman, L.; Anderson, S.M.; Maller, J.L.; Miller, W.T.; Edwards, D.P. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell 2001, 8, 269–280. [Google Scholar] [CrossRef]
- Shimizu, A.; Maruyama, T.; Tamaki, K.; Uchida, H.; Asada, H.; Yoshimura, Y. Impairment of decidualization in SRC-deficient mice. Biol. Reprod. 2005, 73, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kannan, A.; DeMayo, F.J.; Lydon, J.P.; Cooke, P.S.; Yamagishi, H.; Srivastava, D.; Bagchi, M.K.; Bagchi, I.C. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 2011, 331, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Takamoto, N.; Zhao, B.; Tsai, S.Y.; DeMayo, F.J. Identification of Indian hedgehog as a progesterone-responsive gene in the murine uterus. Mol. Endocrinol. 2002, 16, 2338–2348. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Zhao, X.; Das, S.K.; Hogan, B.L.; Dey, S.K. Indian hedgehog as a progesterone-responsive factor mediating epithelial-mesenchymal interactions in the mouse uterus. Dev. Biol. 2002, 245, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, I.; Lee, D.K.; Petit, F.G.; Jeong, J.; Lee, K.; Lydon, J.P.; DeMayo, F.J.; Tsai, M.J.; Tsai, S.Y. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007, 3, e102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kodithuwakku, S.P.; Ng, P.Y.; Chai, J.; Ng, E.H.; Yeung, W.S.; Ho, P.C.; Lee, K.F. Excessive ovarian stimulation up-regulates the Wnt-signaling molecule DKK1 in human endometrium and may affect implantation: An in vitro co-culture study. Hum. Reprod. 2010, 25, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kannan, A.; Das, A.; Demayo, F.J.; Hornsby, P.J.; Young, S.L.; Taylor, R.N.; Bagchi, M.K.; Bagchi, I.C. WNT4 acts downstream of BMP2 and functions via beta-catenin signaling pathway to regulate human endometrial stromal cell differentiation. Endocrinology 2013, 154, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Lu, Z.; Goto, T.; Fusi, L.; Higham, J.; Francis, J.; Withey, A.; Hardt, J.; Cloke, B.; Stavropoulou, A.V.; et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol. Endocrinol. 2007, 21, 2334–2349. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Zhao, Y.B.; Pan, J.L.; Lei, Y.; Liu, M.; Long, Y.; Zhang, J.H.; Hu, Y.; Xu, M.Q.; Yuan, D.Z.; et al. Progesterone-Induced miR-152 Inhibits the Proliferation of Endometrial Epithelial Cells by Downregulating WNT-1. Reprod. Sci. 2017, 24, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Ma, L.; Ma, W.G.; Maas, R.L.; Dey, S.K. Hoxa-10 regulates uterine stromal cell responsiveness to progesterone during implantation and decidualization in the mouse. Mol. Endocrinol. 1999, 13, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Fazleabas, A.T. Uterine receptivity and implantation: The regulation and action of insulin-like growth factor binding protein-1 (IGFBP-1), HOXA10 and forkhead transcription factor-1 (FOXO-1) in the baboon endometrium. Reprod. Biol. Endocrinol. 2004, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buser, A.C.; Gass-Handel, E.K.; Wyszomierski, S.L.; Doppler, W.; Leonhardt, S.A.; Schaack, J.; Rosen, J.M.; Watkin, H.; Anderson, S.M.; Edwards, D.P. Progesterone receptor repression of prolactin/signal transducer and activator of transcription 5-mediated transcription of the beta-casein gene in mammary epithelial cells. Mol. Endocrinol. 2007, 21, 106–125. [Google Scholar] [CrossRef] [PubMed]
- Wieser, F.; Schneeberger, C.; Hudelist, G.; Singer, C.; Kurz, C.; Nagele, F.; Gruber, C.; Huber, J.C.; Tschugguel, W. Endometrial nuclear receptor co-factors SRC-1 and N-CoR are increased in human endometrium during menstruation. Mol. Hum. Reprod. 2002, 8, 644–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, C.W.; Wilson, E.M.; Apparao, K.B.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2002, 87, 2960–2966. [Google Scholar] [CrossRef] [PubMed]
- Pru, J.K.; Clark, N.C. PGRMC1 and PGRMC2 in uterine physiology and disease. Front. Neurosci. 2013, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.S.; Pierron, V.; Michalovich, D.; Astle, S.; Thornton, S.; Peltoketo, H.; Lam, E.W.; Gellersen, B.; Huhtaniemi, I.; Allen, J.; et al. Regulated expression of putative membrane progestin receptor homologues in human endometrium and gestational tissues. J. Endocrinol. 2005, 187, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsano, S.; Quinonero, A.; Perez, S.; Garrido Gomez, T.; Simon, C.; Dominguez, F. Dynamic expression of PGRMC1 and SERBP1 in human endometrium: An implication in the human decidualization process. Fertil. Steril. 2017, 108, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gomez, T.; Quinonero, A.; Antunez, O.; Diaz-Gimeno, P.; Bellver, J.; Simon, C.; Dominguez, F. Deciphering the proteomic signature of human endometrial receptivity. Hum. Reprod. 2014, 29, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Karteris, E.; Zervou, S.; Pang, Y.; Dong, J.; Hillhouse, E.W.; Randeva, H.S.; Thomas, P. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: Potential role in functional progesterone withdrawal at term. Mol. Endocrinol. 2006, 20, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.; Pang, Y.; Thomas, P.; Zhu, Y. Cell-surface expression, progestin binding, and rapid nongenomic signaling of zebrafish membrane progestin receptors α and beta in transfected cells. J. Endocrinol. 2006, 190, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Vigano, P.; Mangioni, S.; Pompei, F.; Chiodo, I. Maternal-conceptus cross talk—A review. Placenta 2003, 24 (Suppl. B), S56–S61. [Google Scholar] [CrossRef]
- Patel, B.; Elguero, S.; Thakore, S.; Dahoud, W.; Bedaiwy, M.; Mesiano, S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Hum. Reprod. Update 2015, 21, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Halasz, M.; Szekeres-Bartho, J. The role of progesterone in implantation and trophoblast invasion. J. Reprod. Immunol. 2013, 97, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florio, P.; Rossi, M.; Vigano, P.; Luisi, S.; Torricelli, M.; Torres, P.B.; Di Blasio, A.M.; Petraglia, F. Interleukin 1beta and progesterone stimulate activin a expression and secretion from cultured human endometrial stromal cells. Reprod. Sci. 2007, 14, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Wetendorf, M.; Wu, S.P.; Wang, X.; Creighton, C.J.; Wang, T.; Lanz, R.B.; Blok, L.; Tsai, S.Y.; Tsai, M.J.; Lydon, J.P.; et al. Decreased epithelial progesterone receptor A at the window of receptivity is required for preparation of the endometrium for embryo attachment. Biol. Reprod. 2017, 96, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jessmon, P.; Leach, R.E.; Armant, D.R. Diverse functions of HBEGF during pregnancy. Mol. Reprod. Dev. 2009, 76, 1116–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Miyake, K.; Voit, R.; Nemoto, S.; Wakeland, E.K.; Grummt, I.; Miyazaki, T. Death-effector domain-containing protein DEDD is an inhibitor of mitotic Cdk1/cyclin B1. Proc. Natl. Acad. Sci. USA 2007, 104, 2289–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Kitazume, M.; Ose, R.; Kurokawa, J.; Koga, K.; Osuga, Y.; Arai, S.; Miyazaki, T. Death effector domain-containing protein (DEDD) is required for uterine decidualization during early pregnancy in mice. J. Clin. Investig. 2011, 121, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Gao, F.; Rusie, A.; Hemingway, J.; Ostmann, A.B.; Sroga, J.M.; Jegga, A.G.; Das, S.K. Decidual cell polyploidization necessitates mitochondrial activity. PLoS ONE 2011, 6, e26774. [Google Scholar] [CrossRef] [PubMed]
- Margarit, L.; Taylor, A.; Roberts, M.H.; Hopkins, L.; Davies, C.; Brenton, A.G.; Conlan, R.S.; Bunkheila, A.; Joels, L.; White, J.O.; et al. MUC1 as a discriminator between endometrium from fertile and infertile patients with PCOS and endometriosis. J. Clin. Endocrinol. Metab. 2010, 95, 5320–5329. [Google Scholar] [CrossRef] [PubMed]
- Nikas, G.; Makrigiannakis, A. Endometrial pinopodes and uterine receptivity. Ann. N. Y. Acad. Sci. 2003, 997, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Mercader, A.; Gimeno, M.J.; Pellicer, A. The interleukin-1 system and human implantation. Am. J. Reprod. Immunol. 1997, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Paulesu, L.; Romagnoli, R.; Bigliardi, E. Materno-fetal immunotolerance: Is Interleukin-1 a fundamental mediator in placental viviparity? Dev. Comp. Immunol. 2005, 29, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.P.; Hamilton, A.E.; Talbi, S.; Dosiou, C.; Nyegaard, M.; Nayak, N.; Genbecev-Krtolica, O.; Mavrogianis, P.; Ferrer, K.; Kruessel, J.; et al. Decidual stromal cell response to paracrine signals from the trophoblast: Amplification of immune and angiogenic modulators. Biol. Reprod. 2007, 76, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Staun-Ram, E.; Shalev, E. Human trophoblast function during the implantation process. Reprod. Biol. Endocrinol. 2005, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Szekeres-Bartho, J.; Par, G.; Dombay, G.; Smart, Y.C.; Volgyi, Z. The antiabortive effect of progesterone-induced blocking factor in mice is manifested by modulating NK activity. Cell. Immunol. 1997, 177, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Par, G.; Geli, J.; Kozma, N.; Varga, P.; Szekeres-Bartho, J. Progesterone regulates IL12 expression in pregnancy lymphocytes by inhibiting phospholipase A2. Am. J. Reprod. Immunol. 2003, 49, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Faust, Z.; Laskarin, G.; Rukavina, D.; Szekeres-Bartho, J. Progesterone-induced blocking factor inhibits degranulation of natural killer cells. Am. J. Reprod. Immunol. 1999, 42, 71–75. [Google Scholar] [PubMed]
- Blois, S.M.; Ilarregui, J.M.; Tometten, M.; Garcia, M.; Orsal, A.S.; Cordo-Russo, R.; Toscano, M.A.; Bianco, G.A.; Kobelt, P.; Handjiski, B.; et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat. Med. 2007, 13, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Sonderegger, S.; Pollheimer, J.; Knofler, M. Wnt signalling in implantation, decidualisation and placental differentiation—Review. Placenta 2010, 31, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.A.; Jonnaert, M.; Labelle-Dumais, C.; Kuroda, K.; Clarke, H.J.; Dufort, D. Uterine Wnt/beta-catenin signaling is required for implantation. Proc. Natl. Acad. Sci. USA 2005, 102, 8579–8584. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Erikson, D.W.; Tilford, S.A.; Bany, B.M.; Maclean, J.A., 2nd; Rucker, E.B., 3rd; Johnson, G.A.; Spencer, T.E. Wnt genes in the mouse uterus: Potential regulation of implantation. Biol. Reprod. 2009, 80, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tranguch, S.; Jia, X.; Zhang, H.; Das, S.K.; Dey, S.K.; Kuo, C.J.; Wang, H. Inactivation of nuclear Wnt-beta-catenin signaling limits blastocyst competency for implantation. Development 2008, 135, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, J.; Zhang, S.; Wang, S.; Wang, W.; Wang, B.; Wang, F.; Chen, Q.; Duan, E.; Leitges, M.; et al. Wnt6 is essential for stromal cell proliferation during decidualization in mice. Biol. Reprod. 2013, 88, 5. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmae, S.; Koel, M.; Vosa, U.; Adler, P.; Suhorutsenko, M.; Laisk-Podar, T.; Kukushkina, V.; Saare, M.; Velthut-Meikas, A.; Krjutskov, K.; et al. Meta-signature of human endometrial receptivity: A meta-analysis and validation study of transcriptomic biomarkers. Sci. Rep. 2017, 7, 10077. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Nguyen, H.P.; Elgass, K.; Simpson, R.J.; Salamonsen, L.A. Human Endometrial Exosomes Contain Hormone-Specific Cargo Modulating Trophoblast Adhesive Capacity: Insights into Endometrial-Embryo Interactions. Biol. Reprod. 2016, 94, 38. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Carver, J.; Ridley, A.J.; Mardon, H.J. Human endometrial stromal cell rho GTPases have opposing roles in regulating focal adhesion turnover and embryo invasion in vitro. Biol. Reprod. 2010, 83, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Neufeld, J.; Reimann, K.; Wittmann, S.; Samalecos, A.; Wolf, A.; Bamberger, A.M.; Gellersen, B. Expansion of human trophoblastic spheroids is promoted by decidualized endometrial stromal cells and enhanced by heparin-binding epidermal growth factor-like growth factor and interleukin-1 beta. Mol. Hum. Reprod. 2011, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Reimann, K.; Samalecos, A.; Aupers, S.; Bamberger, A.M. Invasiveness of human endometrial stromal cells is promoted by decidualization and by trophoblast-derived signals. Hum. Reprod. 2010, 25, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenke, M.; Knofler, M.; Velicky, P.; Weimar, C.H.; Kruse, M.; Samalecos, A.; Wolf, A.; Macklon, N.S.; Bamberger, A.M.; Gellersen, B. Control of human endometrial stromal cell motility by PDGF-BB, HB-EGF and trophoblast-secreted factors. PLoS ONE 2013, 8, e54336. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, R.P.; Lambalk, C.B.; Huirne, J.; Mijatovic, V.; Repping, S.; Hamer, G.; Mastenbroek, S. High-quality human preimplantation embryos actively influence endometrial stromal cell migration. J. Assist. Reprod. Genet. 2018, 35, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Carver, J.G.; Ridley, A.J.; Mardon, H.J. Implantation of the human embryo requires Rac1-dependent endometrial stromal cell migration. Proc. Natl. Acad. Sci. USA 2008, 105, 16189–16194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollheimer, J.; Loregger, T.; Sonderegger, S.; Saleh, L.; Bauer, S.; Bilban, M.; Czerwenka, K.; Husslein, P.; Knofler, M. Activation of the canonical wingless/T-cell factor signaling pathway promotes invasive differentiation of human trophoblast. Am. J. Pathol. 2006, 168, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, L.M.; Chakraborty, C.; McKinnon, T.; Lala, P.K. Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through α 5 beta 1 integrin via mitogen-activated protein Kinase pathway. J. Clin. Endocrinol. Metab. 2001, 86, 2484–2493. [Google Scholar] [PubMed]
- Haouzi, D.; Dechaud, H.; Assou, S.; Monzo, C.; de Vos, J.; Hamamah, S. Transcriptome analysis reveals dialogues between human trophectoderm and endometrial cells during the implantation period. Hum. Reprod. 2011, 26, 1440–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawada, K.; Upadhyay, G.; Ferandon, S.; Janarthanan, S.; Hall, M.; Vilardaga, J.P.; Yajnik, V. Cell migration is regulated by platelet-derived growth factor receptor endocytosis. Mol. Cell Biol. 2009, 29, 4508–4518. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Liu, Y.Q.; Zhou, W.H.; Zhang, Y.Z. Trophoblast-derived chemokine CXCL12 promotes CXCR4 expression and invasion of human first-trimester decidual stromal cells. Hum. Reprod. 2012, 27, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Briese, J.; Oberndorfer, M.; Redlin, K.; Samalecos, A.; Richter, D.U.; Loning, T.; Schulte, H.M.; Bamberger, A.M. Expression of the metastasis suppressor KAI1 in decidual cells at the human maternal-fetal interface: Regulation and functional implications. Am. J. Pathol. 2007, 170, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Wolf, A.; Kruse, M.; Schwenke, M.; Bamberger, A.M. Human endometrial stromal cell-trophoblast interactions: Mutual stimulation of chemotactic migration and promigratory roles of cell surface molecules CD82 and CEACAM1. Biol. Reprod. 2013, 88, 80. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Sheehan, P.M.; Brennecke, S.P.; Keogh, R.J. Vessel remodelling, pregnancy hormones and extravillous trophoblast function. Mol. Cell. Endocrinol. 2012, 349, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Kanzaki, H.; Nakayama, H.; Fujimoto, M.; Hatayama, H.; Kojima, K.; Iwai, M.; Mori, T.; Fujita, J. Induction of tissue inhibitor of metalloproteinase 3 gene expression during in vitro decidualization of human endometrial stromal cells. Endocrinology 1995, 136, 4973–4981. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Lovett, D.H.; Shalev, E. Mechanisms of matrix metalloproteinase-2 (mmp-2) transcriptional repression by progesterone in jar choriocarcinoma cells. Reprod. Biol. Endocrinol. 2009, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Wissink, S.; van Heerde, E.C.; vand der Burg, B.; van der Saag, P.T. A dual mechanism mediates repression of NF-kappaB activity by glucocorticoids. Mol. Endocrinol. 1998, 12, 355–363. [Google Scholar] [PubMed]
- Bruner, K.L.; Rodgers, W.H.; Gold, L.I.; Korc, M.; Hargrove, J.T.; Matrisian, L.M.; Osteen, K.G. Transforming growth factor beta mediates the progesterone suppression of an epithelial metalloproteinase by adjacent stroma in the human endometrium. Proc. Natl. Acad. Sci. USA 1995, 92, 7362–7366. [Google Scholar] [CrossRef] [PubMed]
- Schroen, D.J.; Brinckerhoff, C.E. Nuclear hormone receptors inhibit matrix metalloproteinase (MMP) gene expression through diverse mechanisms. Gene Exp. 1996, 6, 197–207. [Google Scholar]
- Koshiba, H.; Kitawaki, J.; Ishihara, H.; Kado, N.; Kusuki, I.; Tsukamoto, K.; Honjo, H. Progesterone inhibition of functional leptin receptor mRNA expression in human endometrium. Mol. Hum. Reprod. 2001, 7, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, L.A.; Light, M.M.; Mehrotra, P.; Nowak, R.A. Stimulation of GPR30 increases release of EMMPRIN-containing microvesicles in human uterine epithelial cells. J. Clin. Endocrinol. Metab. 2012, 97, 4613–4622. [Google Scholar] [CrossRef] [PubMed]
- Braundmeier, A.G.; Dayger, C.A.; Mehrotra, P.; Belton, R.J., Jr.; Nowak, R.A. EMMPRIN is secreted by human uterine epithelial cells in microvesicles and stimulates metalloproteinase production by human uterine fibroblast cells. Reprod. Sci. 2012, 19, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Davila, J.; Laws, M.J.; Kannan, A.; Li, Q.; Taylor, R.N.; Bagchi, M.K.; Bagchi, I.C. Rac1 Regulates Endometrial Secretory Function to Control Placental Development. PLoS Genet. 2015, 11, e1005458. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chen, X.; He, B.; Liu, S.; Li, Y.; Wang, Q.; Gao, H.; Wang, S.; Liu, J.; Zhang, S.; et al. ROS are critical for endometrial breakdown via NF-kappaB-COX-2 signaling in a female mouse menstrual-like model. Endocrinology 2014, 155, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Sugino, N.; Karube-Harada, A.; Taketani, T.; Sakata, A.; Nakamura, Y. Withdrawal of ovarian steroids stimulates prostaglandin F2α production through nuclear factor-kappaB activation via oxygen radicals in human endometrial stromal cells: Potential relevance to menstruation. J. Reprod. Dev. 2004, 50, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamonsen, L.A.; Lathbury, L.J. Endometrial leukocytes and menstruation. Hum. Reprod. Update 2000, 6, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, G.; Koga, K.; Nagai, M.; Urata, Y.; Takamura, M.; Harada, M.; Hirata, T.; Hirota, Y.; Ogawa, K.; Inoue, S.; et al. Cyclic stretch augments production of neutrophil chemokines, matrix metalloproteinases, and activin a in human endometrial stromal cells. Am. J. Reprod. Immunol. 2015, 73, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Salamonsen, L.A. In vivo evidence for active matrix metalloproteinases in human endometrium supports their role in tissue breakdown at menstruation. J. Clin. Endocrinol. Metab. 2002, 87, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Schatz, F.; Guzeloglu-Kayisli, O.; Arlier, S.; Kayisli, U.A.; Lockwood, C.J. The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding. Hum. Reprod. Update 2016, 22, 497–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.; Kadir, R.A. Endometrial haemostasis and menstruation. Rev. Endocr. Metab. Disord. 2012, 13, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Chegini, N.; Gold, L.I.; Williams, R.S. Localization of transforming growth factor beta isoforms TGF-beta 1, TGF-beta 2, and TGF-beta 3 in surgically induced endometriosis in the rat. Obstet. Gynecol. 1994, 83, 455–461. [Google Scholar] [PubMed]
- Johnson, M.C.; Torres, M.; Alves, A.; Bacallao, K.; Fuentes, A.; Vega, M.; Boric, M.A. Augmented cell survival in eutopic endometrium from women with endometriosis: Expression of c-myc, TGF-beta1 and bax genes. Reprod. Biol. Endocrinol. 2005, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Casslen, B.; Urano, S.; Ny, T. Progesterone regulation of plasminogen activator inhibitor 1 (PAI-1) antigen and mRNA levels in human endometrial stromal cells. Thromb. Res. 1992, 66, 75–87. [Google Scholar] [CrossRef]
- Schatz, F.; Krikun, G.; Runic, R.; Wang, E.Y.; Hausknecht, V.; Lockwood, C.J. Implications of decidualization-associated protease expression in implantation and menstruation. Semin. Reprod. Endocrinol. 1999, 17, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.R.; Sierra-Rivera, E.; Eisenberg, E.; Osteen, K.G. Progesterone exposure prevents matrix metalloproteinase-3 (MMP-3) stimulation by interleukin-1α in human endometrial stromal cells. J. Clin. Endocrinol. Metab. 2000, 85, 1611–1619. [Google Scholar] [PubMed]
- Pretto, C.M.; Gaide Chevronnay, H.P.; Cornet, P.B.; Galant, C.; Delvaux, D.; Courtoy, P.J.; Marbaix, E.; Henriet, P. Production of interleukin-1α by human endometrial stromal cells is triggered during menses and dysfunctional bleeding and is induced in culture by epithelial interleukin-1α released upon ovarian steroids withdrawal. J. Clin. Endocrinol. Metab. 2008, 93, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Cornet, P.B.; Galant, C.; Eeckhout, Y.; Courtoy, P.J.; Marbaix, E.; Henriet, P. Regulation of matrix metalloproteinase-9/gelatinase B expression and activation by ovarian steroids and LEFTY-A/endometrial bleeding-associated factor in the human endometrium. J. Clin. Endocrinol. Metab. 2005, 90, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Cornet, P.B.; Picquet, C.; Lemoine, P.; Osteen, K.G.; Bruner-Tran, K.L.; Tabibzadeh, S.; Courtoy, P.J.; Eeckhout, Y.; Marbaix, E.; Henriet, P. Regulation and function of LEFTY-A/EBAF in the human endometrium. mRNA expression during the menstrual cycle, control by progesterone, and effect on matrix metalloprotineases. J. Biol. Chem. 2002, 277, 42496–42504. [Google Scholar] [CrossRef] [PubMed]
- Salamonsen, L.A.; Woolley, D.E. Menstruation: Induction by matrix metalloproteinases and inflammatory cells. J. Reprod. Immunol. 1999, 44, 1–27. [Google Scholar] [CrossRef]
- Collett, G.P.; Kohnen, G.; Campbell, S.; Davenport, A.P.; Jeffers, M.D.; Cameron, I.T. Localization of endothelin receptors in human uterus throughout the menstrual cycle. Mol. Hum. Reprod. 1996, 2, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Philippeaux, M.M.; Piguet, P.F. Expression of tumor necrosis factor-α and its mRNA in the endometrial mucosa during the menstrual cycle. Am. J. Pathol. 1993, 143, 480–486. [Google Scholar] [PubMed]
- Noguchi, Y.; Sato, T.; Hirata, M.; Hara, T.; Ohama, K.; Ito, A. Identification and characterization of extracellular matrix metalloproteinase inducer in human endometrium during the menstrual cycle in vivo and in vitro. J. Clin. Endocrinol. Metab. 2003, 88, 6063–6072. [Google Scholar] [CrossRef] [PubMed]
- Tabibzadeh, S. The signals and molecular pathways involved in human menstruation, a unique process of tissue destruction and remodelling. Mol. Hum. Reprod. 1996, 2, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Tsuzuki, T.; Shindoh, H.; Nishigaki, A.; Yasuda, K.; Kanzaki, H. Regulation of decidualization and angiogenesis in the human endometrium: Mini review. J. Obstet. Gynaecol. Res. 2014, 40, 1180–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krikun, G.; Schatz, F.; Mackman, N.; Guller, S.; Demopoulos, R.; Lockwood, C.J. Regulation of tissue factor gene expression in human endometrium by transcription factors Sp1 and Sp3. Mol. Endocrinol. 2000, 14, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, C.J.; Paidas, M.; Murk, W.K.; Kayisli, U.A.; Gopinath, A.; Huang, S.J.; Krikun, G.; Schatz, F. Involvement of human decidual cell-expressed tissue factor in uterine hemostasis and abruption. Thromb. Res. 2009, 124, 516–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, H.; Spornitz, U.M. Microarchitecture of the human endometrium by scanning electron microscopy: Menstrual desquamation and remodeling. Ann. N. Y. Acad. Sci. 1991, 622, 28–46. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L.; Zhang, L.; Arango, N.A.; Teixeira, J.; Pru, J.K. Mesenchymal-to-epithelial transition contributes to endometrial regeneration following natural and artificial decidualization. Stem Cells Dev. 2013, 22, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Ichim, T.E.; Zhong, J.; Rogers, A.; Yin, Z.; Jackson, J.; Wang, H.; Ge, W.; Bogin, V.; Chan, K.W.; et al. Endometrial regenerative cells: A novel stem cell population. J. Transl. Med. 2007, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousins, F.L.; Gargett, C.E. Endometrial stem/progenitor cells and their role in the pathogenesis of endometriosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Yamagami, K.; Yoshii, Y.; Yamauchi, N. Growth factor induced proliferation, migration, and lumen formation of rat endometrial epithelial cells in vitro. J. Reprod. Dev. 2016, 62, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, J.; Fukaya, T.; Murakami, T.; Yoshida, H.; Yajima, A. Hepatocyte growth factor stimulated proliferation, migration, and lumen formation of human endometrial epithelial cells in vitro. Biol. Reprod. 1997, 57, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.; Yap, J.; Gamage, T.; Salamonsen, L.; Dimitriadis, E.; Menkhorst, E. Galectin-7 is important for normal uterine repair following menstruation. Mol. Hum. Reprod. 2014, 20, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.; D’Sylva, R.; Volpert, M.; Jamsai, D.; Merriner, D.J.; Nie, G.; Salamonsen, L.A.; O’Bryan, M.K. Endometrial CRISP3 is regulated throughout the mouse estrous and human menstrual cycle and facilitates adhesion and proliferation of endometrial epithelial cells. Biol. Reprod. 2015, 92, 99. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Kaitu’u-Lino, T.; Salamonsen, L.A. Extracellular matrix dynamics in scar-free endometrial repair: Perspectives from mouse in vivo and human in vitro studies. Biol. Reprod. 2011, 85, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Strug, M.R.; Su, R.W.; Kim, T.H.; Mauriello, A.; Ticconi, C.; Lessey, B.A.; Young, S.L.; Lim, J.M.; Jeong, J.W.; Fazleabas, A.T. RBPJ mediates uterine repair in the mouse and is reduced in women with recurrent pregnancy loss. FASEB J. 2018, 32, 2452–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Krieg, S.; Hwang, J.Y.; Dhal, S.; Kuo, C.J.; Lasley, B.L.; Brenner, R.M.; Nayak, N.R. Dynamic regulation of Wnt7a expression in the primate endometrium: Implications for postmenstrual regeneration and secretory transformation. Endocrinology 2012, 153, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Kaitu’u-Lino, T.J.; Phillips, D.J.; Morison, N.B.; Salamonsen, L.A. A new role for activin in endometrial repair after menses. Endocrinology 2009, 150, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Cousins, F.L.; Kirkwood, P.M.; Murray, A.A.; Collins, F.; Gibson, D.A.; Saunders, P.T. Androgens regulate scarless repair of the endometrial “wound” in a mouse model of menstruation. FASEB J. 2016, 30, 2802–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simitsidellis, I.; Saunders, P.T.K.; Gibson, D.A. Androgens and endometrium: New insights and new targets. Mol. Cell. Endocrinol. 2018, 465, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Kaitu’u-Lino, T.J.; Morison, N.B.; Salamonsen, L.A. Neutrophil depletion retards endometrial repair in a mouse model. Cell Tissue Res. 2007, 328, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [PubMed]
- Arici, A.; Seli, E.; Senturk, L.M.; Gutierrez, L.S.; Oral, E.; Taylor, H.S. Interleukin-8 in the human endometrium. J. Clin. Endocrinol. Metab. 1998, 83, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A.; Okochi, H.; Bradham, D.M.; Grotendorst, G.R. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell. 1993, 4, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002, 5, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Maybin, J.A.; Critchley, H.O. Steroid regulation of menstrual bleeding and endometrial repair. Rev. Endocr. Metab. Disord. 2012, 13, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Aghajanova, L.; Houshdaran, S.; Balayan, S.; Manvelyan, E.; Irwin, J.C.; Huddleston, H.G.; Giudice, L.C. In vitro evidence that platelet-rich plasma stimulates cellular processes involved in endometrial regeneration. J. Assist. Reprod. Genet. 2018, 35, 757–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, C.E.; Chan, R.W.; Schwab, K.E. Hormone and growth factor signaling in endometrial renewal: Role of stem/progenitor cells. Mol. Cell. Endocrinol. 2008, 288, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, X.; Dai, Y.; Hu, X.; Zhu, H.; Jiang, Y.; Zhang, S. Endometrial stem cells repair injured endometrium and induce angiogenesis via AKT and ERK pathways. Reproduction 2016, 152, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Jiang, Y.; Pan, Y.; Shi, L.; Zhang, S. Human menstrual blood-derived stem cells promote the repair of impaired endometrial stromal cells by activating the p38 MAPK and AKT signaling pathways. Reprod. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Orvis, G.D.; Wang, Y.; Behringer, R.R. Stromal-to-epithelial transition during postpartum endometrial regeneration. PLoS ONE 2012, 7, e44285. [Google Scholar] [CrossRef] [PubMed]
- Suginami, K.; Sato, Y.; Horie, A.; Matsumoto, H.; Tani, H.; Mizumoto, Y.; Ono, M.; Matsuoka, A.; Kyo, S.; Araki, Y.; et al. Platelet-derived microparticles and soluble factors differentially regulate human endometrial epithelial cell movement. Am. J. Reprod. Immunol. 2017, 77, e12641. [Google Scholar] [CrossRef] [PubMed]
- Demir, R.; Yaba, A.; Huppertz, B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2010, 112, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Gargett, C.E.; Rogers, P.A. Human endometrial angiogenesis. Reproduction 2001, 121, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maybin, J.A.; Murray, A.A.; Saunders, P.T.K.; Hirani, N.; Carmeliet, P.; Critchley, H.O.D. Hypoxia and hypoxia inducible factor-1α are required for normal endometrial repair during menstruation. Nat. Commun. 2018, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Greaves, E.; Collins, F.; Critchley, H.O.; Saunders, P.T. ERbeta-dependent effects on uterine endothelial cells are cell specific and mediated via Sp1. Hum. Reprod. 2013, 28, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Chen, Y.J.; Chou, T.Y.; Chen, C.Y.; Li, H.Y.; Huang, B.S.; Tsai, H.W.; Lan, H.Y.; Chang, C.H.; Twu, N.F.; et al. Oestrogen-induced angiogenesis promotes adenomyosis by activating the Slug-VEGF axis in endometrial epithelial cells. J. Cell. Mol. Med. 2014, 18, 1358–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhang, T.; Man, G.C.; May, K.E.; Becker, C.M.; Davis, T.N.; Kung, A.L.; Birsner, A.E.; D’Amato, R.J.; Wong, A.W.; et al. Vascular endothelial growth factor C is increased in endometrium and promotes endothelial functions, vascular permeability and angiogenesis and growth of endometriosis. Angiogenesis 2013, 16, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.J.; Seol, J.W.; Jang, J.Y.; Cho, Y.S.; Kim, K.R.; Choi, Y.; Lydon, J.P.; Demayo, F.J.; Shibuya, M.; et al. VEGF-A regulated by progesterone governs uterine angiogenesis and vascular remodelling during pregnancy. EMBO Mol. Med. 2013, 5, 1415–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.H.; Vlahos, N.; Shih Ie, M.; Zhao, Y. Expression Patterns of VEGF and Flk-1 in Human Endometrium during the Menstrual Cycle. J. Reprod. Infertil. 2015, 16, 3–9. [Google Scholar] [PubMed]
- Fan, X.; Krieg, S.; Kuo, C.J.; Wiegand, S.J.; Rabinovitch, M.; Druzin, M.L.; Brenner, R.M.; Giudice, L.C.; Nayak, N.R. VEGF blockade inhibits angiogenesis and reepithelialization of endometrium. FASEB J. 2008, 22, 3571–3580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharkey, A.M.; Day, K.; McPherson, A.; Malik, S.; Licence, D.; Smith, S.K.; Charnock-Jones, D.S. Vascular endothelial growth factor expression in human endometrium is regulated by hypoxia. J. Clin. Endocrinol. Metab. 2000, 85, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Maybin, J.A.; Hirani, N.; Brown, P.; Jabbour, H.N.; Critchley, H.O. The regulation of vascular endothelial growth factor by hypoxia and prostaglandin F(2)α during human endometrial repair. J. Clin. Endocrinol. Metab. 2011, 96, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Sokkar, P.; Sathis, V.; Ramachandran, M. Computational modeling on the recognition of the HRE motif by HIF-1: Molecular docking and molecular dynamics studies. J. Mol. Model. 2012, 18, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Chen, L.H.; Li, H.Y.; Chang, S.P.; Liao, C.Y.; Tsui, K.H.; Sung, Y.J.; Chao, K.C. Roles of estrogen and progesterone in endometrial hemodynamics and vascular endothelial growth factor production. J. Chin. Med. Assoc. 2009, 72, 188–193. [Google Scholar] [CrossRef]
- Krikun, G.; Schatz, F.; Finlay, T.; Kadner, S.; Mesia, A.; Gerrets, R.; Lockwood, C.J. Expression of angiopoietin-2 by human endometrial endothelial cells: Regulation by hypoxia and inflammation. Biochem. Biophys. Res. Commun. 2000, 275, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Teichert, M.; Milde, L.; Holm, A.; Stanicek, L.; Gengenbacher, N.; Savant, S.; Ruckdeschel, T.; Hasanov, Z.; Srivastava, K.; Hu, J.; et al. Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat. Commun. 2017, 8, 16106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizov, M.; Andreeva, P.; Dimova, I. Molecular regulation and role of angiogenesis in reproduction. Taiwan J. Obstet. Gynecol. 2017, 56, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Okada, H.; Cho, H.; Shimoi, K.; Miyashiro, H.; Yasuda, K.; Kanzaki, H. Divergent regulation of angiopoietin-1, angiopoietin-2, and vascular endothelial growth factor by hypoxia and female sex steroids in human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 168, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, A.; Okada, H.; Okamoto, R.; Sugiyama, S.; Miyazaki, K.; Yasuda, K.; Kanzaki, H. Concentrations of stromal cell-derived factor-1 and vascular endothelial growth factor in relation to the diameter of human follicles. Fertil. Steril. 2011, 95, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, A.; Okada, H.; Nakamoto, T.; Okamoto, R.; Yasuda, K.; Kanzaki, H. Estrogen induces stromal cell-derived factor 1 (SDF-1/CXCL12) production in human endometrial stromal cells: A possible role of endometrial epithelial cell growth. Fertil. Steril. 2011, 95, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Glace, L.; Grygielko, E.T.; Boyle, R.; Wang, Q.; Laping, N.J.; Sulpizio, A.C.; Bray, J.D. Estrogen-induced stromal cell-derived factor-1 (SDF-1/Cxcl12) expression is repressed by progesterone and by Selective Estrogen Receptor Modulators via estrogen receptor α in rat uterine cells and tissues. Steroids 2009, 74, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Osuga, Y.; Tsutsumi, O.; Yano, T.; Yoshino, O.; Takai, Y.; Matsumi, H.; Hiroi, H.; Kugu, K.; Momoeda, M.; et al. Demonstration of angiogenin in human endometrium and its enhanced expression in endometrial tissues in the secretory phase and the decidua. J. Clin. Endocrinol. Metab. 2001, 86, 5609–5614. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.E.; Fraser, R.; Gurung, S.; Goulwara, S.S.; Whitley, G.S.; Johnstone, A.P.; Cartwright, J.E. Increased angiogenic factor secretion by decidual natural killer cells from pregnancies with high uterine artery resistance alters trophoblast function. Hum. Reprod. 2014, 29, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lash, G.E.; Pitman, H.; Morgan, H.L.; Innes, B.A.; Agwu, C.N.; Bulmer, J.N. Decidual macrophages: Key regulators of vascular remodeling in human pregnancy. J. Leukoc. Biol. 2016, 100, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choi, Y.M.; Chae, H.D.; Kim, C.H.; Kang, B.M. Decreased expression of angiogenin in the eutopic endometrium from women with advanced stage endometriosis. J. Korean Med. Sci. 2008, 23, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.C.; Bicknell, R. Angiogenesis in the endometrium. Angiogenesis 1998, 2, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Lecce, G.; Meduri, G.; Ancelin, M.; Bergeron, C.; Perrot-Applanat, M. Presence of estrogen receptor beta in the human endometrium through the cycle: Expression in glandular, stromal, and vascular cells. J. Clin. Endocrinol. Metab. 2001, 86, 1379–1386. [Google Scholar] [PubMed]
- Krikun, G.; Schatz, F.; Taylor, R.; Critchley, H.O.; Rogers, P.A.; Huang, J.; Lockwood, C.J. Endometrial endothelial cell steroid receptor expression and steroid effects on gene expression. J. Clin. Endocrinol. Metab. 2005, 90, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Critchley, H.O.; Kelly, R.W.; Brenner, R.M.; Baird, D.T. The endocrinology of menstruation—A role for the immune system. Clin. Endocrinol. (Oxf.) 2001, 55, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.H.; Yoon, J.Y.; Lee, S.H.; Bryja, V.; Andersson, E.R.; Arenas, E.; Kwon, Y.G.; Choi, K.Y. Wnt5a is required for endothelial differentiation of embryonic stem cells and vascularization via pathways involving both Wnt/beta-catenin and protein kinase Cα. Circ. Res. 2009, 104, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.M.; Kitajewski, J.; D’Amore, P.A. Wnt1 and Wnt5a affect endothelial proliferation and capillary length; Wnt2 does not. Growth Factors 2007, 25, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, E.; Vago, R.; Sanchez, A.M.; Podini, P.; Zarovni, N.; Murdica, V.; Rizzo, R.; Bortolotti, D.; Candiani, M.; Viganò, P. Secretome of in vitro cultured human embryos contains extracellular vesicles that are uptaken by the maternal side. Sci. Rep. 2017, 7, 5210. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makieva, S.; Giacomini, E.; Ottolina, J.; Sanchez, A.M.; Papaleo, E.; Viganò, P. Inside the Endometrial Cell Signaling Subway: Mind the Gap(s). Int. J. Mol. Sci. 2018, 19, 2477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092477
Makieva S, Giacomini E, Ottolina J, Sanchez AM, Papaleo E, Viganò P. Inside the Endometrial Cell Signaling Subway: Mind the Gap(s). International Journal of Molecular Sciences. 2018; 19(9):2477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092477
Chicago/Turabian StyleMakieva, Sofia, Elisa Giacomini, Jessica Ottolina, Ana Maria Sanchez, Enrico Papaleo, and Paola Viganò. 2018. "Inside the Endometrial Cell Signaling Subway: Mind the Gap(s)" International Journal of Molecular Sciences 19, no. 9: 2477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092477