Normal and Abortive Buds Transcriptomic Profiling of Broccoli ogu Cytoplasmic Male Sterile Line and Its Maintainer

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. High-Throughput Transcriptome Sequencing and Unigene Assembly
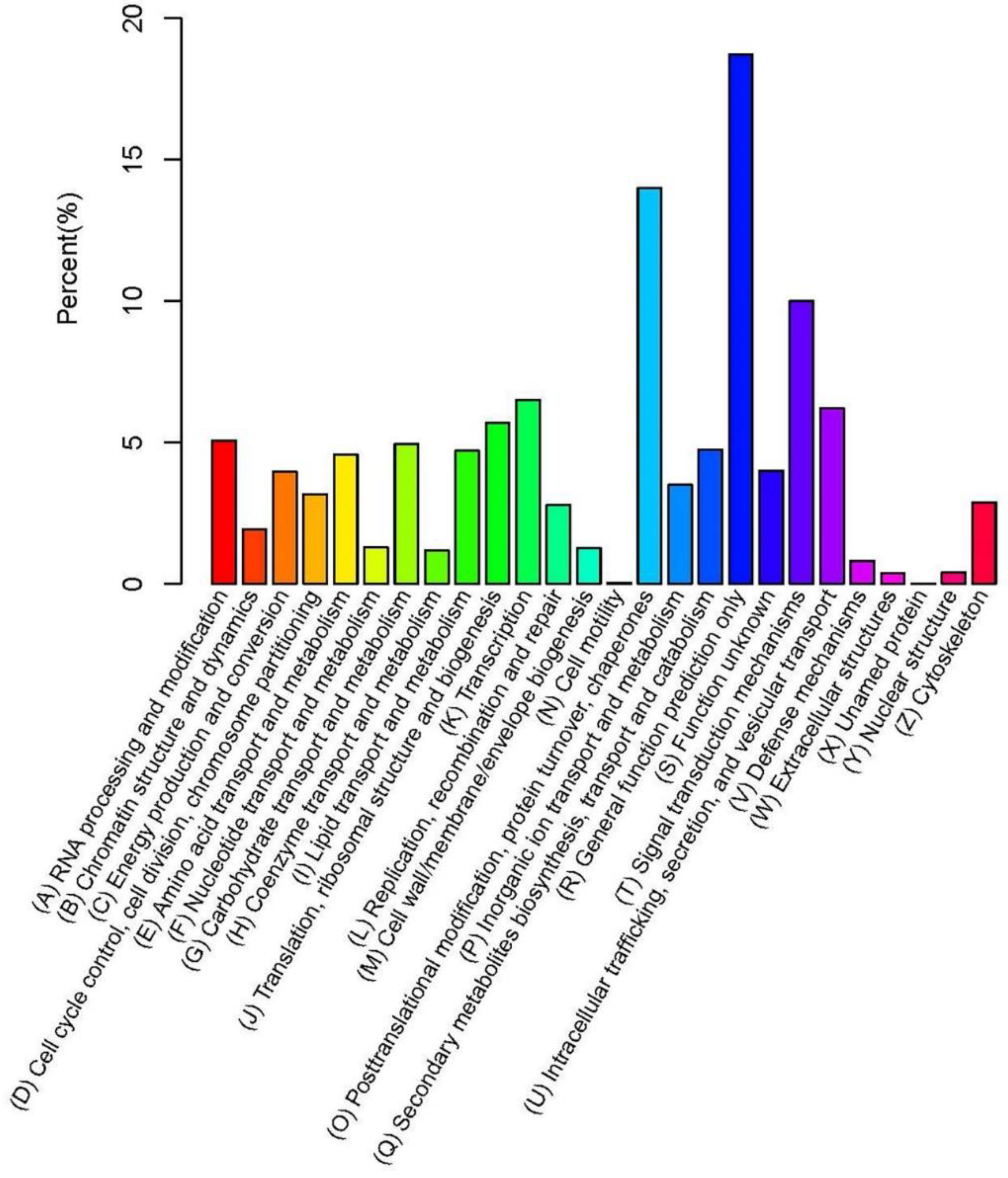
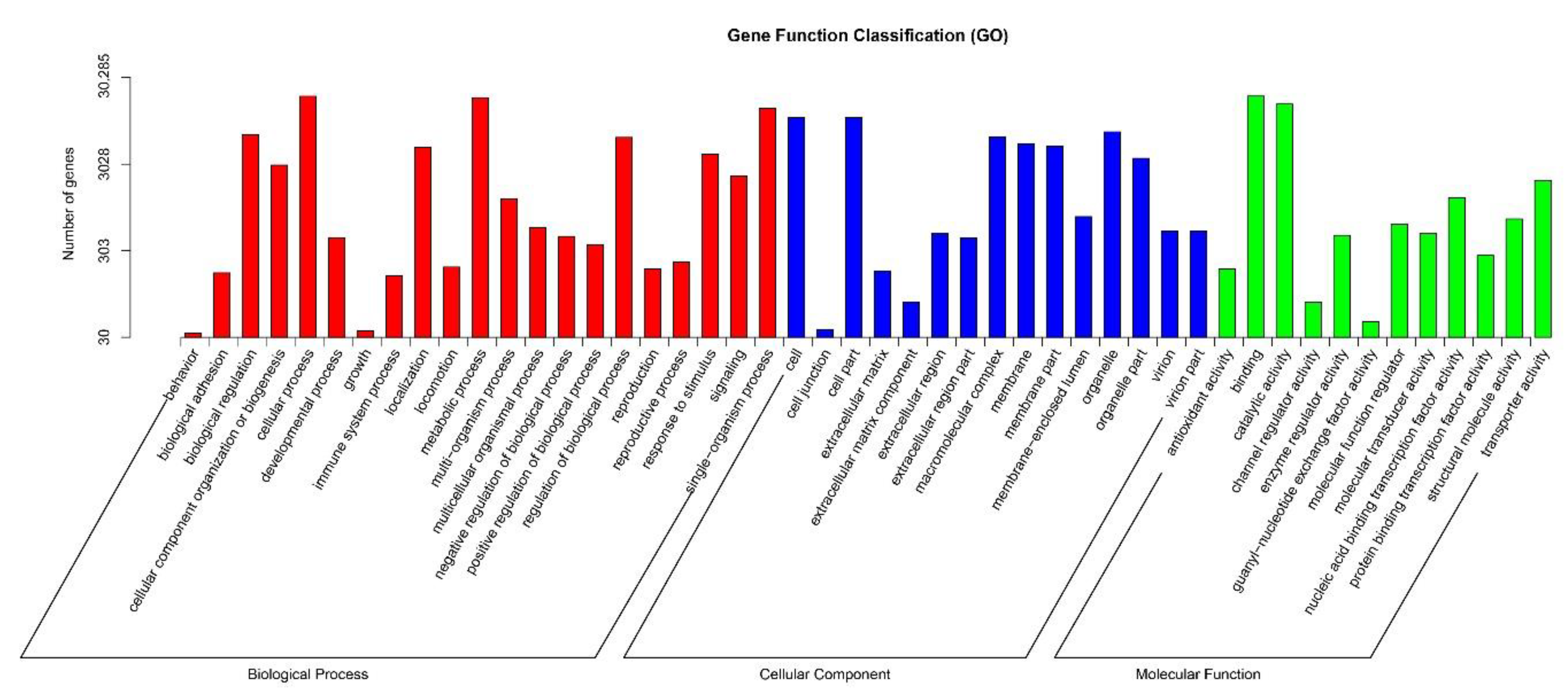
2.2. Gene Annotation and Functional Classification
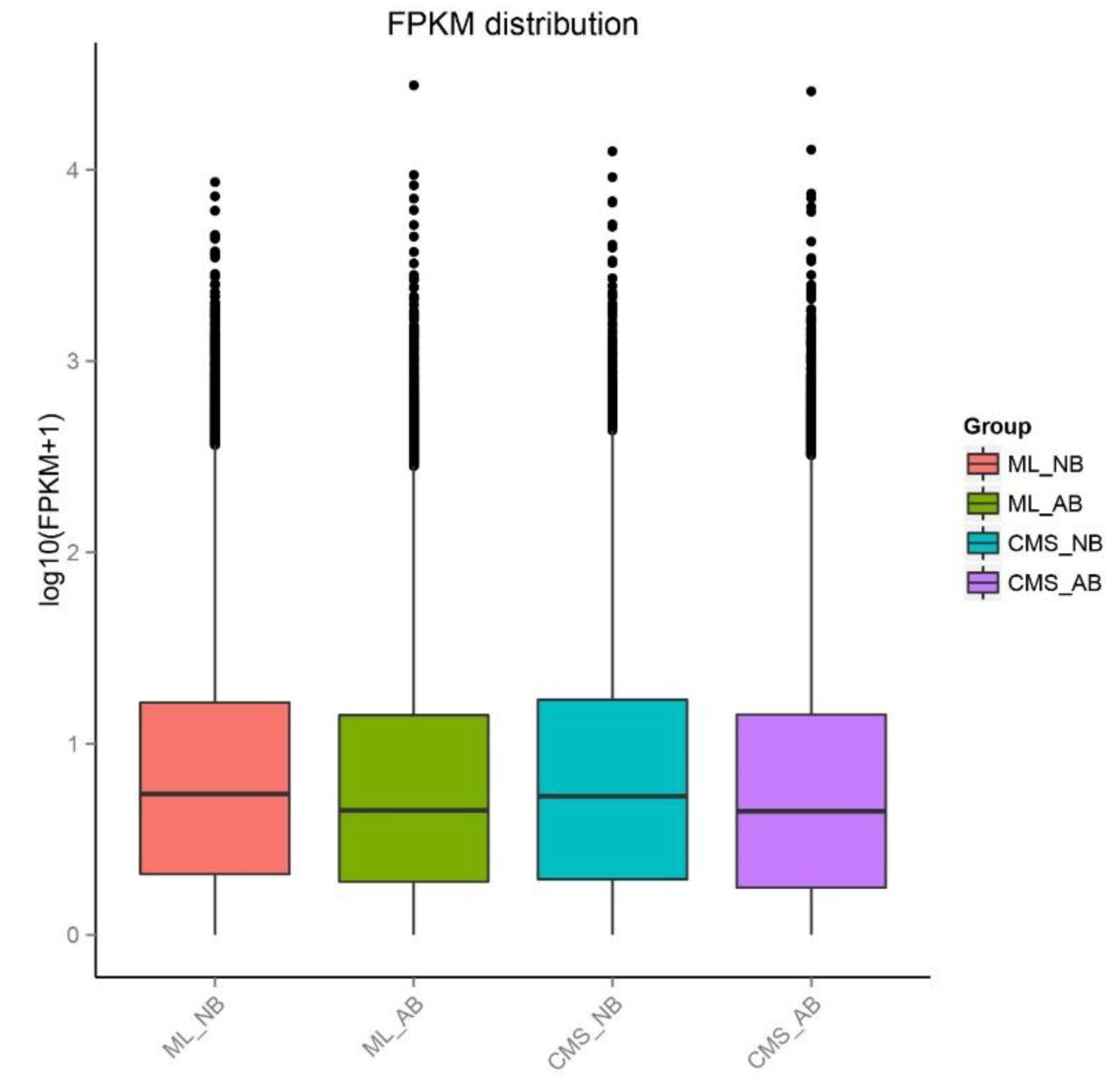
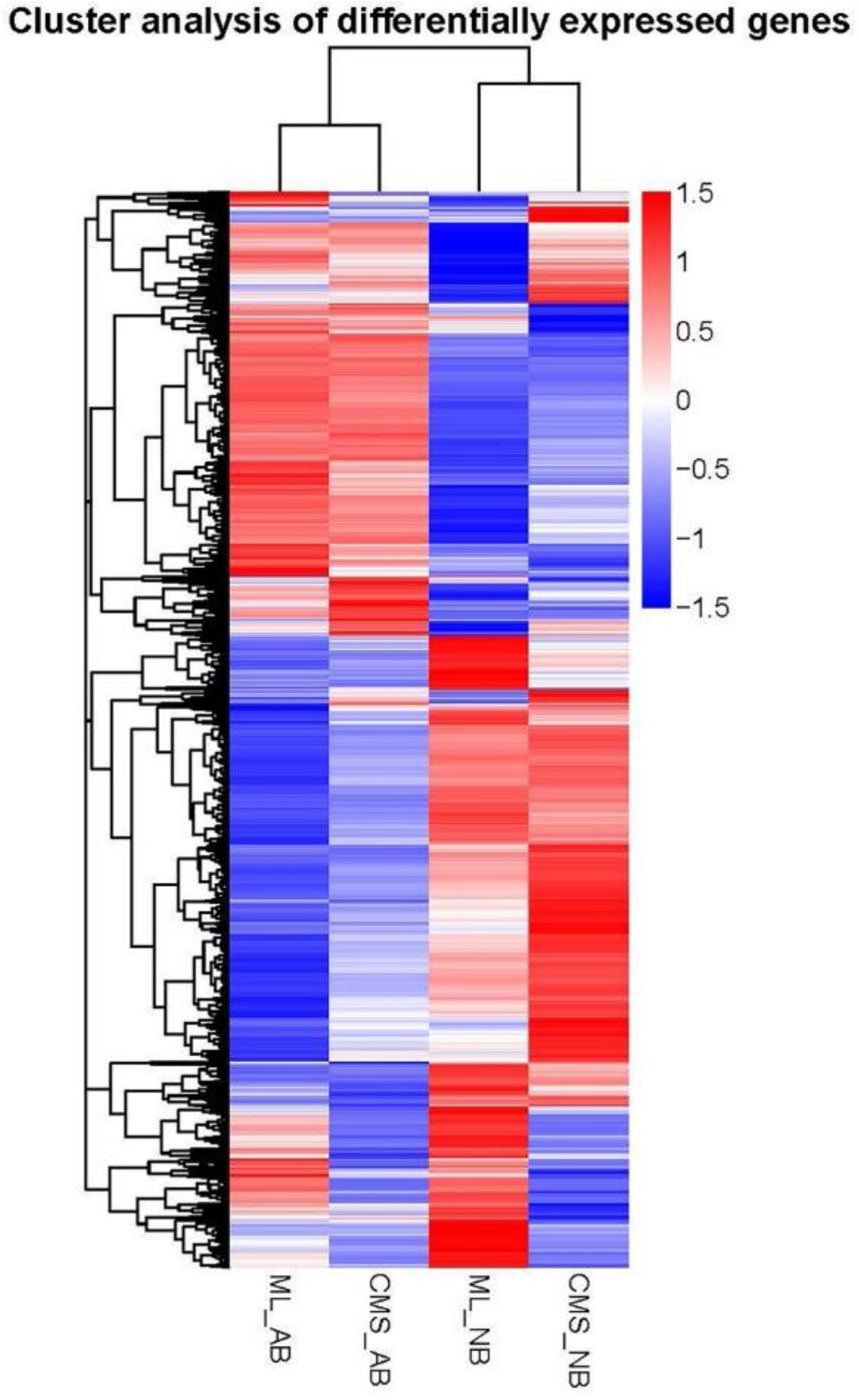
2.3. Genes Express Differences and DEGs Clustering
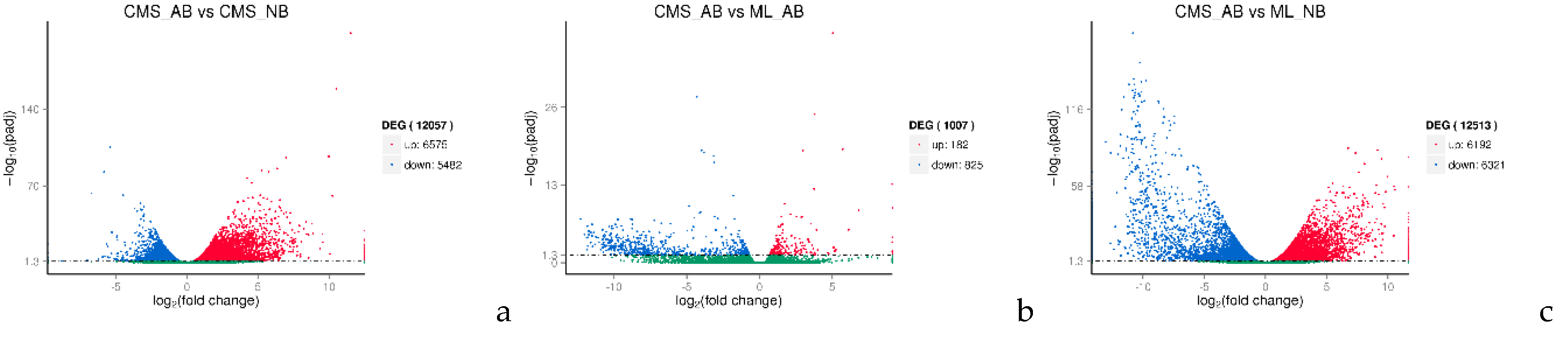
2.4. DEGs in Normal and Abortive Buds of ogu CMS
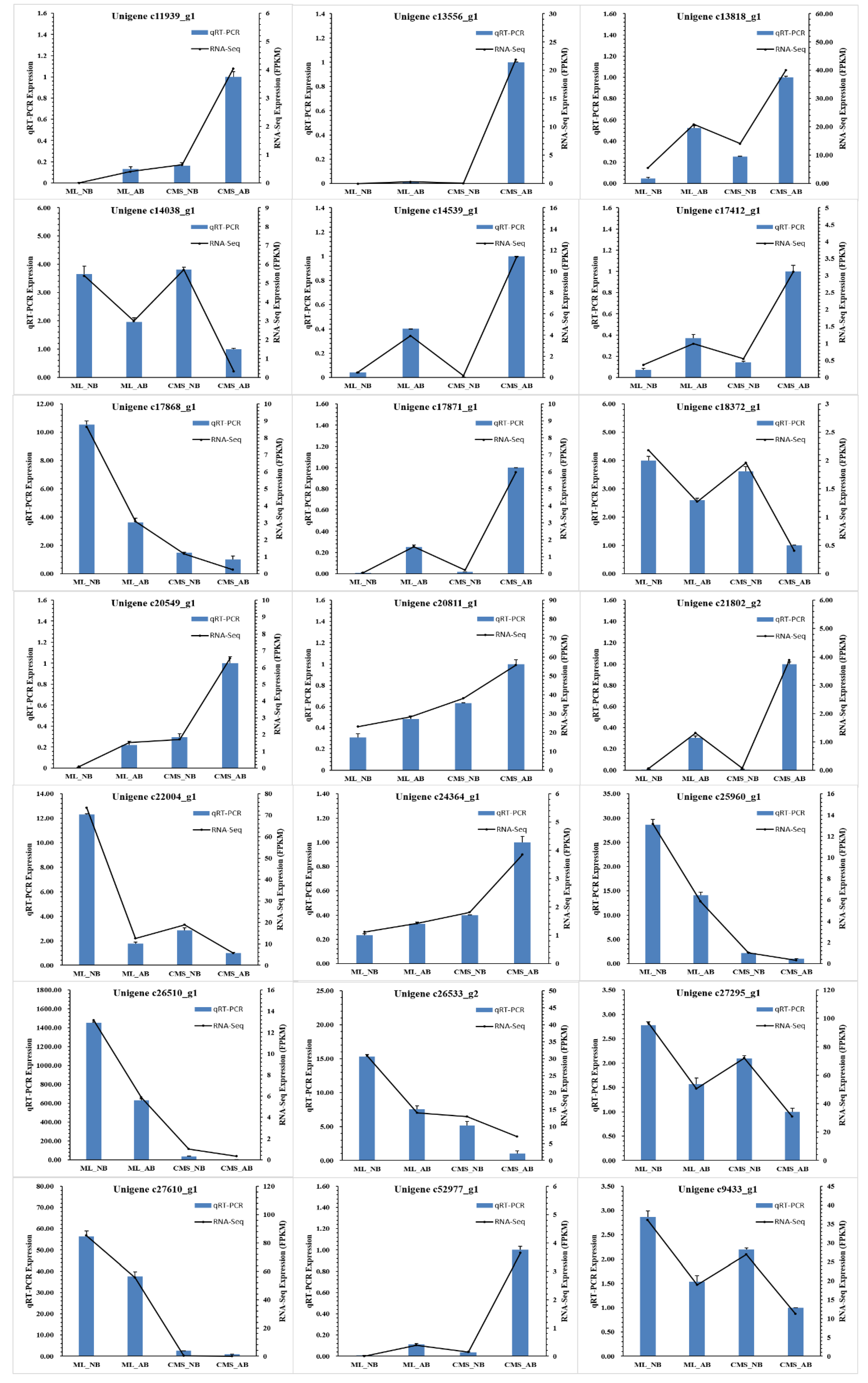
2.5. Validation of RNA-Seq Data by Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
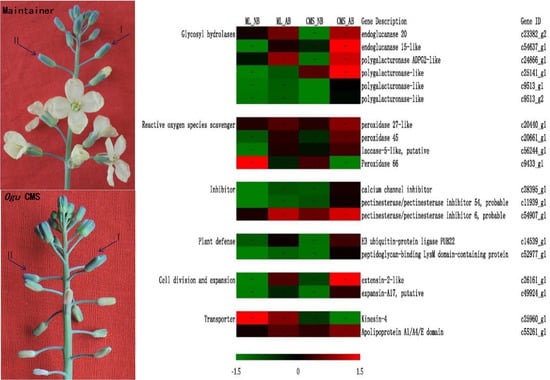
2.6. Ogu CMS Bud Abortion-Related Genes in Broccoli
2.7. Transcription Factors Are Involved in Broccoli ogu CMS Bud Abortion Control
3. Discussion
3.1. Genes Related to Programmed Cell Death Are Involved in Bud Abortion
3.2. Glycosyl Hydrolases, Inhibitors and Plant Defence Related Genes Are Implicated in Bud Abortion
3.3. Transcription Factors Associated with Bud Development
3.4. Molecular Mechanisms Associated with CMS
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Quality Testing
4.3. RNA-Seq Library Construction and Illumina Sequencing
4.4. RNA-Seq Data Quality Control and Transcriptome de Novo Assembly
4.5. Unigene Function Annotation
4.6. Analysis of DEGs
4.7. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Li, Q.F.; Huang, W.W.; Xu, X.Y.; Zhang, X.L.; Hui, M.X.; Zhang, M.K.; Zhang, L.G. A vacuolar processing enzyme RsVPE1 gene of radish is involved in floral bud abortion under heat stress. Int. J. Mol. Sci. 2013, 14, 13346–13359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, X.L.; Zhang, L.G.; Hui, M.X.; Zhang, M.K. Analysis of differential gene expression during floral bud abortion in radish (Raphanus sativus L.). Genet. Mol. Res. 2013, 12, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, L.G.; Wang, F.M.; Hui, M.X.; Zhang, M.K. Observation of histocytological feature on radish flower bud during aborting. Acta Agric. Bor. Occid. Sin. 2008, 5, 272–276. [Google Scholar]
- Wang, Q.B.; Zhang, Y.Y.; Liu, Y.M.; Yang, L.M.; Zhuang, M.; Sun, P.T. The floral and seed setting characteristics in two types of male sterile lines of cabbage (Brassica oleracea L. var. capitata). Acta Hortic. Sin. 2011, 38, 61–68. [Google Scholar]
- Shu, J.S.; Liu, Y.M.; Li, Z.S.; Zhang, L.L.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Zhang, Y.Y.; Li, Z.S.; Sun, P.T. Study on the floral characteristics and structure in two types of male sterile lines of broccoli (Brassica oleracea var. italica). J. Plant. Genet. Resour. 2014, 15, 113–119. [Google Scholar]
- Jia, J.; Zhang, L.G. mRNA differential display and EST sequence analysis of aborted bud and normal bud in radish (Raphanus sativus). Acta Agric. Nucl. Sin. 2008, 22, 426–431. [Google Scholar]
- Zhang, L.G.; Jia, J.; Zhang, S.L.; Zhang, Y. cDNA-AFLP differential expression analysis of genes related with aborting bud in Chinese cabbage. J. Agric. Biotechnol. 2010, 18, 489–492. [Google Scholar]
- Wang, Q.; Liu, Z.; Feng, H. cDNA-AFLP differential expression analysis of genes about bud aborting in genetic male sterile line of Brassica campestris L. ssp. chinensis (L.) Makino var. rosularis Tsen et Lee. Mol. Plant. Breed. 2014, 12, 118–126. [Google Scholar]
- Kensler, T.W.; Chen, J.G.; Egner, P.A.; Fahey, J.W.; Jacobson, L.P.; Stephenson, K.K.; Ye, L.X.; Coady, J.L.; Wang, J.B.; Wu, Y.; et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Canene-Adams, K.; Lindshield, B.L.; Wang, S.; Jeffery, E.H.; Clinton, S.K.; Erdman, J.W., Jr. Combinations of tomato and broccoli enhance antitumor activity in dunning r3327-h prostate adenocarcinomas. Cancer Res. 2007, 67, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, E.H.; Araya, M. Physiological effects of broccoli consumption. Phytochem. Rev. 2009, 8, 283–298. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [Green Version]
- Thomas, S.G.; Franklin-Tong, V.E. Self-incompatibility triggers programmed cell death in Papaver pollen. Nature 2004, 429, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Reape, T.J.; Molony, E.M.; McCabe, P.F. Programmed cell death in plants: Distinguishing between different modes. J. Exp. Bot. 2008, 59, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Rybaczek, D.; Musiałek, M.W.; Balcerczyk, A. Caffeine-induced premature chromosome condensation results in the apoptosis-like programmed cell death in root meristems of Vicia faba. PLoS ONE 2015, 10, e0142307. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Franklin-Tong, V.E. Temporal and spatial activation of caspase-like enzymes induced by self-incompatibility in Papaver pollen. Proc. Natl. Acad. Sci. USA 2007, 104, 18327–18332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, K.A.; Bancroft, J.; Bosch, M.; Ings, J.; Smirnoff, N.; Franklin-Tong, V.E. Reactive oxygen species and nitric oxide mediate actin reorganization and programmed cell death in the self-incompatibility response of papaver. Plant Physiol. 2011, 156, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.J.; Wang, Z.Q.; Dong, W.; Sun, C.Q.; Wang, H.B.; Song, A.P.; He, L.Z.; Fang, W.M.; Chen, F.D.; Teng, N.J. Transcriptomic and proteomic analysis reveals mechanisms of embryo abortion during chrysanthemum cross breeding. Sci. Rep. 2014, 4, 6536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagundes, D.; Bohn, B.; Cabreira, C.; Leipelt, F.; Dias, N.; Bodanese-Zanettini, M.H.; Cagliari, A. Caspases in plants: Metacaspase gene family in plant stress responses. Funct. Integr. Genom. 2015, 15, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Carranza, Z.H.; Elliott, K.A.; Roberts, J.A. Expression of polygalacturonases and evidence to support their role during cell separation processes in Arabidopsis thaliana. J. Exp. Bot. 2007, 58, 3719–3730. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Kay, P.; Wilson, S.; Swain, S.M. ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell 2009, 21, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Libertini, E.; Li, Y.; McQueen-Mason, S.J. Phylogenetic analysis of the plant endo-β-1, 4-glucanase gene family. J. Mol. Evol. 2004, 58, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Kawano, T.; Kadono, T.; Fumoto, K.; Lapeyrie, F.; Kuse, M.; Isobe, M.; Furuichi, T.; Muto, S. Aluminum as a specific inhibitor of plant TPC1 Ca2+ channels. Biochem. Biophys. Res. Commun. 2004, 324, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Peiter, E.; Maathuis, F.J.; Mills, L.N.; Knight, H.; Pelloux, J.; Hetherington, A.M.; Sanders, D. The vacuolar Ca2+-activated channel TPC1 regulates germination and stomatal movement. Nature 2005, 434, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.W.; Wang, J.L.; An, L.L.; Doerge, R.W.; Chen, Z.J.; Grau, C.R.; Meng, J.L.; Osborn, T.C. Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 2007, 227, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Wan, J.; Czechowski, T.; Udvardi, M.; Stacey, G. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol. Plant Microbe Interact. 2007, 20, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.H.; Ryu, M.Y.; Jammes, F.; Hwang, J.H.; Turek, M.; Kang, B.G.; Kwak, J.M.; Kim, W.T. Roles of four Arabidopsis U-Box E3 ubiquitin ligases in negative regulation of abscisic acid-mediated drought stress responses. Plant Physiol. 2012, 160, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.R.; Zhang, X.C.; Neece, D.; Ramonell, K.M.; Clough, S.; Kim, S.Y.; Stacey, M.G.; Stacey, G. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 2008, 20, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Brotmana, Y.; Landau, U.; Pnini, S.; Lisec, J.; Balazadeh, S.; Mueller-Roeber, B.; Zilberstein, A.; Willmitzer, L.; Chet, I.; Viterbo, A. The LysM receptor-like kinase LysM RLK1 is required to activate defense and abiotic-stress responses induced by overexpression of fungal chitinases in Arabidopsis plants. Mol. Plant. 2012, 5, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nguyen, C.T.; Liang, Y.; Cao, Y.; Stacey, G. Role of LysM receptors in chitin-triggered plant innate immunity. Plant. Signal. Behav. 2013, 8, e22598. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.J.; Lucena, C.; Romera, F.J.; Alcantara, E.; Perez-Vicente, R. Ethylene and nitric oxide involvement in the up-regulation of key genes related to iron acquisition and homeostasis in Arabidopsis. J. Exp. Bot. 2010, 61, 3885–3899. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.S.; Chen, M.; Li, L.C.; Ma, Y.Z. Functions and application of the AP2/ERF transcription factor family in crop improvement. J. Integr. Plant Biol. 2011, 53, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Shpak, E.D. ERECTA family genes regulate development of cotyledons during embryogenesis. FEBS Lett. 2014, 588, 3912–3917. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Fujisawa, M.; Shima, Y.; Ito, Y. The AP2/ERF transcription factor SlERF52 functions in flower pedicel abscission in tomato. J. Exp. Bot. 2014, 65, 3111–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Nakanot, T. Development and regulation of pedicel abscission in tomato. Front. Plant Sci. 2015, 6, 442. [Google Scholar] [CrossRef] [PubMed]
- Jisha, V.; Dampanaboina, L.; Vadassery, J.; Mithöfer, A.; Kappara, S.; Ramanan, R. Overexpression of an AP2/ERF type transcription factor OsEREBP1 confers biotic and abiotic stress tolerance in rice. PLoS ONE 2015, 10, e0127831. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Phukan, U.J.; Tripathi, V.; Singh, D.K.; Luqman, S.; Shukla, R.K. PsAP2 an AP2/ERF family transcription factor from Papaver somniferum enhances abiotic and biotic stress tolerance in transgenic tobacco. Plant Mol. Biol. 2015, 89, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Babitha, K.C.; Vemanna, R.S.; Nataraja, K.N.; Udayakumar, M. Overexpression of EcbHLH57 transcription factor from Eleusine coracana L. in tobacco confers tolerance to salt, oxidative and drought stress. PLoS ONE 2015, 10, e0137098. [Google Scholar] [CrossRef] [PubMed]
- Heyman, J.; Cools, T.; Vandenbussche, F.; Heyndrickx, K.S.; Van Leene, J.; Vercauteren, I.; Vanderauwera, S.; Vandepoele, K.; De Jaeger, G.; Van Der, S.D.; et al. ERF115 controls root quiescent center cell division and stem cell replenishment. Science 2013, 342, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix–loop–helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Brownfield, L.; Khatab, H.; Sidorova, A.; Lingaya, M.; Twel, D. The R2R3 MYB transcription factor DUO1 activates a male germline-specific regulon essential for sperm cell differentiation in Arabidopsis. Plant Cell 2011, 23, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Aeschbacher, R.A.; Schrott, M.; Potrykus, I.; Saul, M.W. Isolation and molecular characterization of PosF21, an Arabidopsis thaliana gene which shows characteristics of a b-Zip class transcription factor. Plant J. 1991, 1, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.H.; Zhu, Q.; Dai, S.H.; Lamb, C.; Beachy, R.N. RF2a, a bZIP transcriptional activator of the phloem-specific rice tungro bacilliform virus promoter, functions in vascular development. EMBO J. 1997, 16, 5247–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Cao, K.; Wang, X. A conserved proline residue in the leucine zipper region of AtbZIP34 and AtbZIP61 in Arabidopsis thaliana interferes with the formation of homodimer. Biochem. Biophys. Res. Commun. 2007, 362, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Gibalová, A.; Renák, D.; Matczuk, K.; Dupl’áková, N.; Cháb, D.; Twell, D.; Hongys, D. AtbZIP34 is required for Arabidopsis pollen wall patterning and the control of several metabolic pathways in developing pollen. Plant Mol. Biol. 2009, 70, 581–601. [Google Scholar]
- Yang, P.; Han, J.; Huang, J. Transcriptome sequencing and de novo analysis of cytoplasmic male sterility and maintenance in JA-CMS cotton. PLoS ONE 2014, 9, e112320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, X.; Li, S.; Hu, G.; Zhang, L.; Song, X. Tapetal-Delayed programmed cell death (PCD) and oxidative stress-induced male sterility of Aegilops uniaristata cytoplasm in wheat. Int. J. Mol. Sci. 2018, 19, 1708. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, S.; Ding, X.; He, T.; Dai, J.; Yang, S.; Gai, J. Comparative transcriptome analysis between the cytoplasmic male sterile line NJCMS1A and its maintainer NJCMS1B in soybean (Glycine max (L.) Merr.). PLoS ONE 2015, 10, e0126771. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Li, A.; Wang, Y. Morphological structure and transcriptome comparison of the cytoplasmic male sterility line in Brassica napus (SaNa-1A) derived from somatic hybridization and its maintainer line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, X.F.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lan, Y.; Wen, C.; Zhao, H.; Wang, J.; Wang, Y. Transcriptome sequencing analyses between the cytoplasmic male sterile line and its maintainer line in Welsh onion (Allium fistulosum L.). Int. J. Mol. Sci. 2016, 17, 1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ye, J.; Jia, Y.; Zhang, L.; Song, X. ITRAQ-based proteomics analyses of sterile/fertile anthers from a thermo-sensitive cytoplasmic male-sterile wheat with Aegilops kotschyi cytoplasm. Int. J. Mol. Sci. 2018, 19, 1344. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cheng, J.; Qin, C.; Hu, Z.; Yin, C.; Hu, K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int. J. Mol. Sci. 2013, 14, 22982–22996. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Liu, T.; Wang, Z. Comparative transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int. J. Mol. Sci. 2016, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Liu, Y.; Li, Z.; Zhang, L.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Organelle simple sequence repeat markers help to distinguish carpelloid stamen and normal cytoplasmic male sterile sources in broccoli. PLoS ONE 2015, 10, e0138750. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wood, W.I. A profile hidden Markov model for signal peptides generated by HMMER. Bioinformatics 2003, 19, 307–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Nat. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Wallenius, K.T. Biased Sampling: The Non-Central Hypegeometric Probability Distribution. Ph.D. Thesis, Stanford University, Stanford, CA, USA, 1963. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-Seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lobato, M.E.; Hasperuéb, J.H.; Civelloa, P.M.; Chaves, A.R.; Martínez, G.A. Effect of 1-MCP on the expression of chlorophyll degrading genes during senescence of broccoli (Brassica oleracea L.). Sci. Hortic 2012, 144, 208–211. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
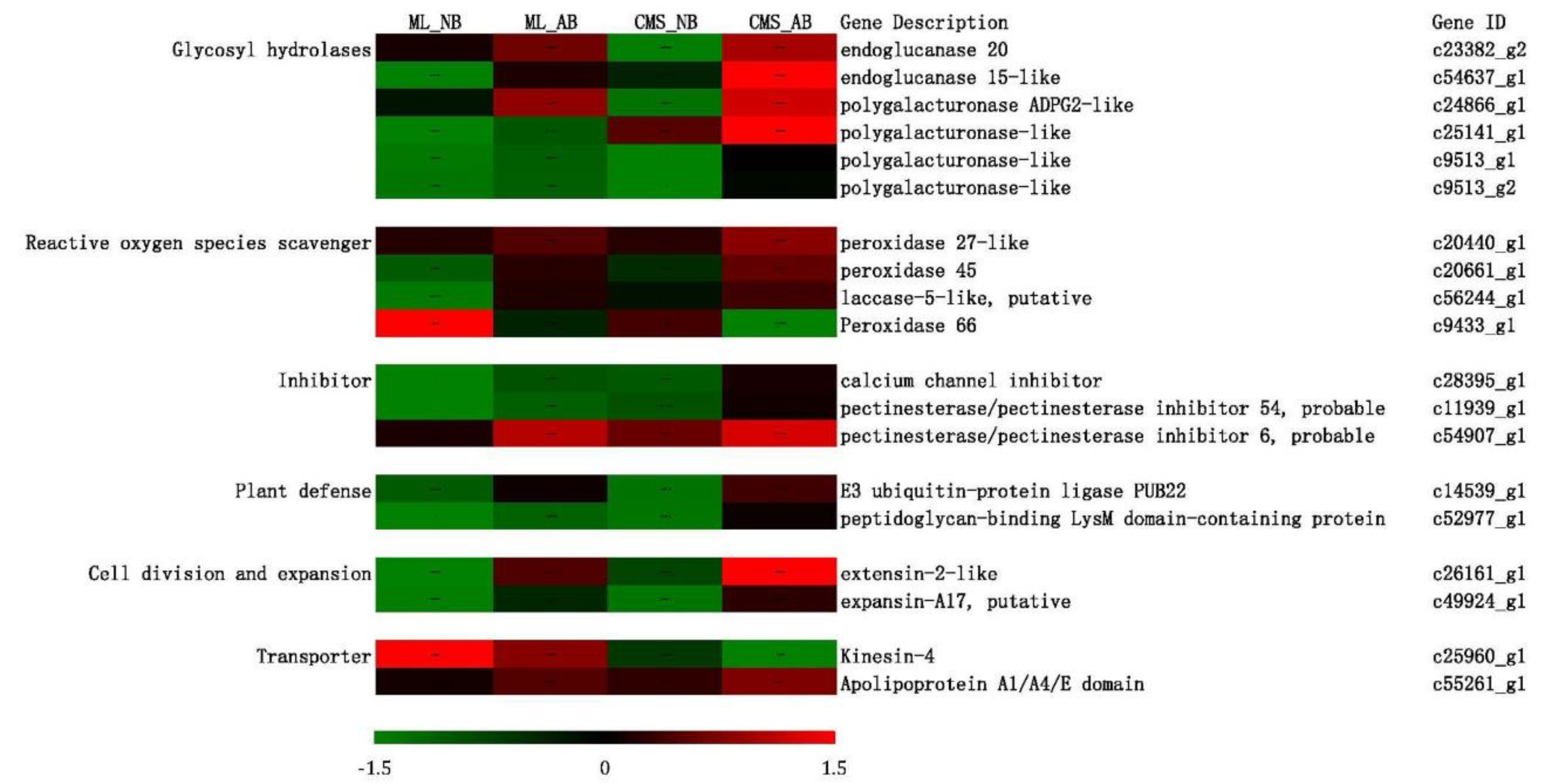{kind=link}
| Sample Name | Raw Reads | Clean Reads (Clean/Raw) | Clean Bases (Gb) | Q20 (%) | GC Content (%) | Mapped Reads (Mapped/Clean) |
|---|---|---|---|---|---|---|
| ML_NB1 | 57,767,246 | 55,194,896 (95.55%) | 8.28 | 94.30 | 46.93 | 41,252,694 (74.74%) |
| ML_NB2 | 44,577,570 | 43,849,450 (98.37%) | 6.58 | 92.93 | 46.46 | 32,614,176 (74.38%) |
| ML_NB3 | 61,251,852 | 59,738,162(97.53%) | 8.96 | 94.03 | 46.96 | 44,511,290 (74.51%) |
| ML_AB1 | 61,588,740 | 60,493,582 (98.22%) | 9.08 | 94.32 | 46.73 | 45,965,016 (75.98%) |
| ML_AB2 | 54,823,836 | 53,530,342 (97.64%) | 8.02 | 93.90 | 46.74 | 40,115,268 (74.94%) |
| ML_AB3 | 58,528,852 | 56,475,844 (96.49%) | 8.48 | 94.33 | 46.81 | 42,816,816 (75.81%) |
| CMS_NB1 | 54,455,258 | 53,170,054 (97.64%) | 7.98 | 94.23 | 47.02 | 39,776,946 (74.81%) |
| CMS_NB2 | 53,265,514 | 52,284,140 (98.16%) | 7.84 | 93.97 | 47.12 | 38,965,364 (74.53%) |
| CMS_NB3 | 51,816,268 | 50,884,086 (98.20%) | 7.64 | 93.79 | 46.91 | 37,800,172 (74.29%) |
| CMS_AB1 | 64,340,176 | 63,146,694 (98.15%) | 9.48 | 94.16 | 47.04 | 47,846,088 (75.77%) |
| CMS_AB2 | 56,566,280 | 55,530,456 (98.17%) | 8.32 | 94.27 | 46.78 | 42,582,890 (76.68%) |
| CMS_AB3 | 60,701,770 | 59,404,212 (97.86%) | 8.92 | 94.09 | 46.70 | 44,549,934 (74.99%) |
| Length | 200–500 bp | 500–1 kbp | 1 k–2 kbp | >2 kbp | Total | Min (bp) | Mean (bp) | Median (bp) | Max (bp) | N50 | Total Nucleotides |
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of transcripts | 44,030 | 21,348 | 21,467 | 10,502 | 97,347 | 201 | 936 | 578 | 16,361 | 1510 | 91,137,323 |
| No. of Unigenes | 37,344 | 12,545 | 10,691 | 5470 | 66,050 | 201 | 786 | 424 | 16,361 | 1363 | 51,896,834 |
| Sequence Database | Number of Annotated Unigenes | Percentage of Annotated Unigene Sequences (%) | Software and Parameters |
|---|---|---|---|
| Annotated in Nr | 44,294 | 67.06 | NCBI blast 2.2.28+, e-value = 1 × 10−5 |
| Annotated in Nt | 50,157 | 75.93 | NCBI blast 2.2.28+, e-value = 1 × 10−5 |
| Annotated in KO | 12,403 | 18.77 | KAAS, KEGG Automatic Annotation Server, e-value = 1 × 10−10 |
| Annotated in SwissProt | 29,861 | 45.2 | NCBI blast 2.2.28+, e-value = 1 × 10−5 |
| Annotated in PFAM | 24,660 | 37.33 | HMMER 3.0 package, hmmscan, e-value = 0.01 |
| Annotated in GO | 30,285 | 45.85 | Blast2GO v2.5 [12] and self-write the script, e-value = 1 × 10−6 |
| Annotated in KOG | 12,492 | 18.91 | NCBI blast 2.2.28+, e-value = 1 × 10−3 |
| Annotated in all Databases | 6070 | 9.19 | - |
| Annotated in at least one Database | 54,753 | 82.89 | - |
| Total Unigenes | 66,050 | 100 | - |
| GO Accession | Description | Term Type | p Value | Corrected p Value | DEG Item | Gene Names |
|---|---|---|---|---|---|---|
| GO:0071555 | cell wall organization | Biological process | 9.34 × 10−10 | 3.25 × 10−6 | 9 | c23382_g2, c9513_g2, c9513_g1, c24866_g1, c25141_g1, c11939_g1, c54907_g1, c49924_g1, c26161_g1 |
| GO:0045229 | external encapsulating structure organization | Biological process | 1.34 × 10−9 | 3.25 × 10−6 | 9 | c54907_g1, c26161_g1, c49924_g1, c23382_g2, c24866_g1, c25141_g1, c11939_g1, c9513_g1, c9513_g2 |
| GO:0071554 | cell wall organization or biogenesis | Biological process | 1.66 × 10−9 | 3.25 × 10−6 | 10 | c49924_g1, c26161_g1, c52977_g1, c54907_g1, c9513_g1, c9513_g2, c24866_g1, c25141_g1, c11939_g1, c23382_g2 |
| GO:0005985 | sucrose metabolic process | Biological process | 6.13 × 10−6 | 0.005404 | 8 | c54907_g1, c54637_g1, c23382_g2, c25141_g1, c11939_g1, c24866_g1, c9513_g2, c9513_g1 |
| GO:0005982 | starch metabolic process | Biological process | 6.46 × 10−6 | 0.005404 | 8 | c23382_g2, c25141_g1, c11939_g1, c24866_g1, c9513_g2, c9513_g1, c54907_g1, c54637_g1 |
| GO:0005984 | disaccharide metabolic process | Biological process | 8.99 × 10−6 | 0.005849 | 8 | c54907_g1, c54637_g1, c23382_g2, c9513_g2, c9513_g1, c11939_g1, c25141_g1, c24866_g1 |
| GO:0006073 | cellular glucan metabolic process | Biological process | 1.2 × 10−5 | 0.006395 | 8 | c24866_g1, c11939_g1, c25141_g1, c9513_g2, c9513_g1, c23382_g2, c54637_g1, c54907_g1 |
| GO:0044042 | glucan metabolic process | Biological process | 1.2 × 10−5 | 0.006395 | 8 | c23382_g2, c9513_g1, c9513_g2, c25141_g1, c11939_g1, c24866_g1, c54907_g1, c54637_g1 |
| GO:0009311 | oligosaccharide metabolic process | Biological process | 1.66 × 10−5 | 0.008084 | 8 | c23382_g2, c25141_g1, c11939_g1, c24866_g1, c9513_g2, c9513_g1, c54907_g1, c54637_g1 |
| GO:0044264 | cellular polysaccharide metabolic process | Biological process | 3.33 × 10−5 | 0.013006 | 9 | c54637_g1, c24364_g1, c54907_g1, c24866_g1, c11939_g1, c25141_g1, c9513_g1, c9513_g2, c23382_g2 |
| GO:0005976 | polysaccharide metabolic process | Biological process | 5.27 × 10−5 | 0.017134 | 9 | c25141_g1, c11939_g1, c24866_g1, c9513_g2, c9513_g1, c23382_g2, c54637_g1, c24364_g1, c54907_g1 |
| GO:0044723 | single-organism carbohydrate metabolic process | Biological process | 7.34 × 10−5 | 0.022634 | 11 | c11939_g1, c25141_g1, c24866_g1, c9513_g1, c9513_g2, c562_g1, c23382_g2, c17871_g1, c54637_g1, c54907_g1, c24364_g1 |
| GO:0044262 | cellular carbohydrate metabolic process | Biological process | 0.00015 | 0.043887 | 9 | c23382_g2, c9513_g1, c9513_g2, c25141_g1, c11939_g1, c24866_g1, c24364_g1, c54907_g1, c54637_g1 |
| GO:0030312 | external encapsulating structure | Cellular component | 3.87 × 10−6 | 0.005404 | 6 | c23382_g2, c54907_g1, c49924_g1, c11939_g1, c26161_g1, c24866_g1 |
| GO:0005576 | extracellular region | Cellular component | 7.52 × 10−6 | 0.005507 | 11 | c56244_g1, c28395_g1, c55261_g1, c14539_g1, c49924_g1, c20661_g1, c24866_g1, c25141_g1, c20440_g1, c9513_g2, c9513_g1 |
| GO:0005618 | cell wall | Cellular component | 2.31 × 10−5 | 0.010397 | 5 | c11939_g1, c26161_g1, c24866_g1, c49924_g1, c54907_g1 |
| GO:0071944 | cell periphery | Cellular component | 4.68 × 10−5 | 0.016257 | 8 | c49924_g1, c50518_g1, c26161_g1, c54907_g1, c11939_g1, c24866_g1, c23382_g2, c22601_g2 |
| GO:0004650 | polygalacturonase activity | Molecular function | 6.12 × 10−6 | 0.005404 | 4 | c9513_g1, c9513_g2, c25141_g1, c24866_g1 |
| GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | Molecular function | 2.96 × 10−5 | 0.012366 | 8 | c54637_g1, c25141_g1, c24866_g1, c9513_g1, c9513_g2, c23382_g2, c562_g1, c17871_g1 |
| GO:0016798 | hydrolase activity, acting on glycosyl bonds | Molecular function | 4.72 × 10−5 | 0.016257 | 8 | c17871_g1, c562_g1, c23382_g2, c9513_g1, c9513_g2, c25141_g1, c24866_g1, c54637_g1 |
| #Term | ID | Input Number | p Value | Corrected p-Value | Input |
|---|---|---|---|---|---|
| Phenylalanine metabolism | ko00360 | 3 | 0.000337 | 0.006398357 | c9433_g1, c20661_g1, c20440_g1 |
| Phenylpropanoid biosynthesis | ko00940 | 3 | 0.001398 | 0.013279387 | c9433_g1, c20661_g1, c20440_g1 |
| Propanoate metabolism | ko00640 | 1 | 0.042547 | 0.179678515 | c17871_g1 |
| Cutin, suberine and wax biosynthesis | ko00073 | 1 | 0.048213 | 0.179678515 | c24364_g1 |
| Fatty acid elongation | ko00062 | 1 | 0.051033 | 0.179678515 | c21267_g1 |
| Endocrine and other factor-regulated calcium reabsorption | ko04961 | 1 | 0.060379 | 0.179678515 | c22601_g2 |
| alpha-Linolenic acid metabolism | ko00592 | 1 | 0.083359 | 0.179678515 | c27465_g3 |
| Photosynthesis | ko00195 | 1 | 0.08789 | 0.179678515 | c28693_g1 |
| Synaptic vesicle cycle | ko04721 | 1 | 0.090598 | 0.179678515 | c22601_g2 |
| Parkinson’s disease | ko05012 | 1 | 0.095095 | 0.179678515 | c25708_g1 |
| Glycerolipid metabolism | ko00561 | 1 | 0.104024 | 0.179678515 | c57011_g1 |
| Peroxisome | ko04146 | 1 | 0.132037 | 0.187274571 | c24364_g1 |
| Pyruvate metabolism | ko00620 | 1 | 0.145727 | 0.187274571 | c17871_g1 |
| Cysteine and methionine metabolism | ko00270 | 1 | 0.149117 | 0.187274571 | c17871_g1 |
| Huntington’s disease | ko05016 | 1 | 0.161716 | 0.187274571 | c22601_g2 |
| Glycerophospholipid metabolism | ko00564 | 1 | 0.164214 | 0.187274571 | c57011_g1 |
| Oxidative phosphorylation | ko00190 | 1 | 0.174958 | 0.187274571 | c25708_g1 |
| Glycolysis/Gluconeogenesis | ko00010 | 1 | 0.177418 | 0.187274571 | c17871_g1 |
| Endocytosis | ko04144 | 1 | 0.208772 | 0.208772251 | c22601_g2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, J.; Zhang, L.; Liu, Y.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Normal and Abortive Buds Transcriptomic Profiling of Broccoli ogu Cytoplasmic Male Sterile Line and Its Maintainer. Int. J. Mol. Sci. 2018, 19, 2501. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092501
Shu J, Zhang L, Liu Y, Li Z, Fang Z, Yang L, Zhuang M, Zhang Y, Lv H. Normal and Abortive Buds Transcriptomic Profiling of Broccoli ogu Cytoplasmic Male Sterile Line and Its Maintainer. International Journal of Molecular Sciences. 2018; 19(9):2501. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092501
Chicago/Turabian StyleShu, Jinshuai, Lili Zhang, Yumei Liu, Zhansheng Li, Zhiyuan Fang, Limei Yang, Mu Zhuang, Yangyong Zhang, and Honghao Lv. 2018. "Normal and Abortive Buds Transcriptomic Profiling of Broccoli ogu Cytoplasmic Male Sterile Line and Its Maintainer" International Journal of Molecular Sciences 19, no. 9: 2501. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092501