Telocinobufagin and Marinobufagin Produce Different Effects in LLC-PK1 Cells: A Case of Functional Selectivity of Bufadienolides

 and
and 
Abstract
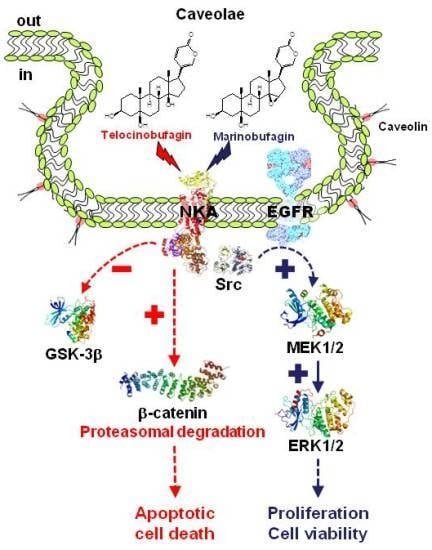
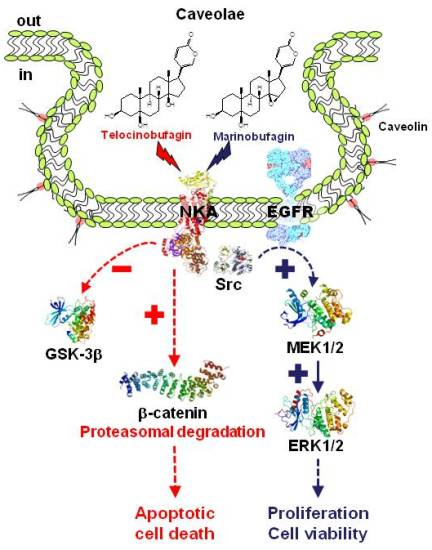
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
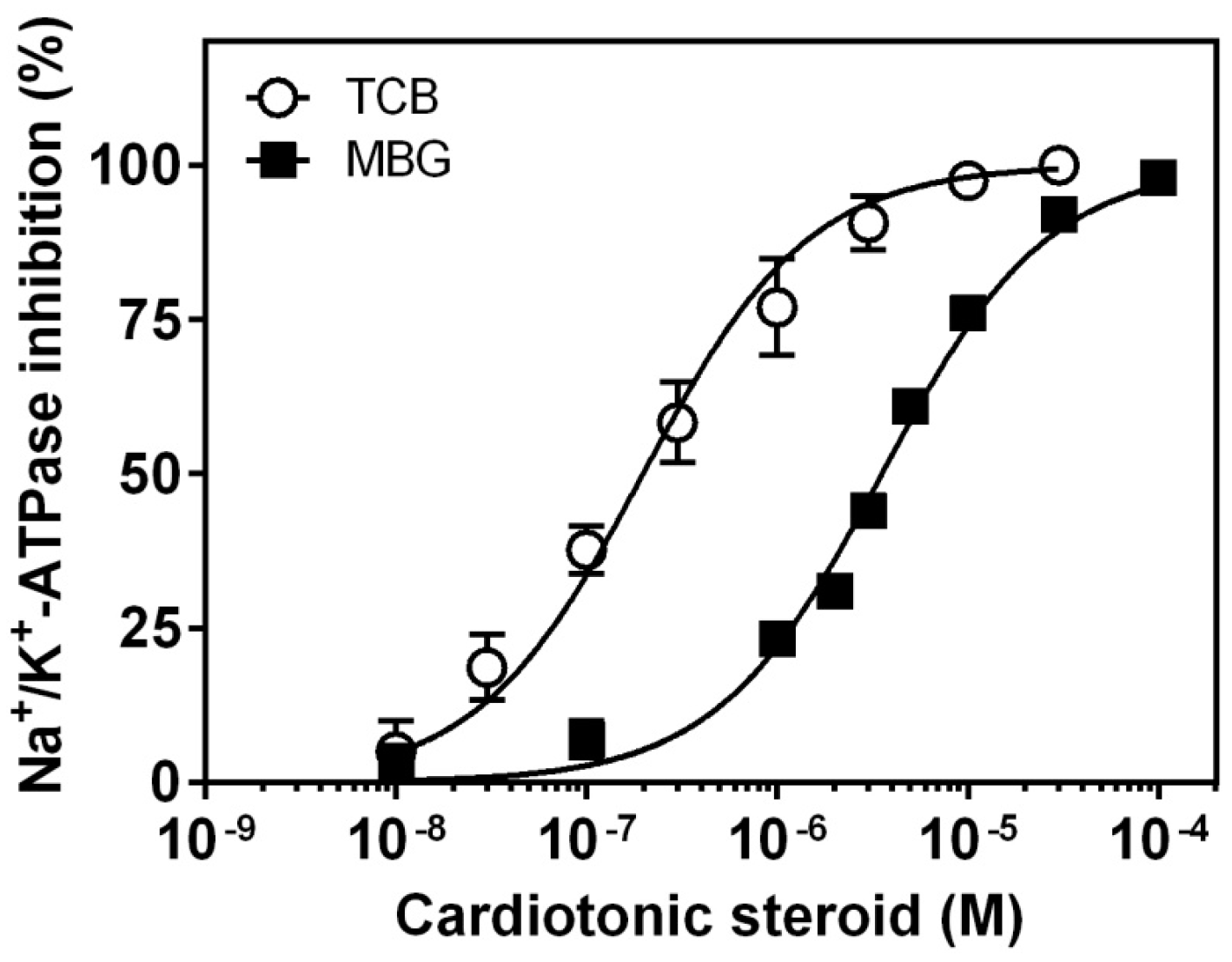
2.1. Inhibition of Pig Kidney Na+/K+-ATPase Activity
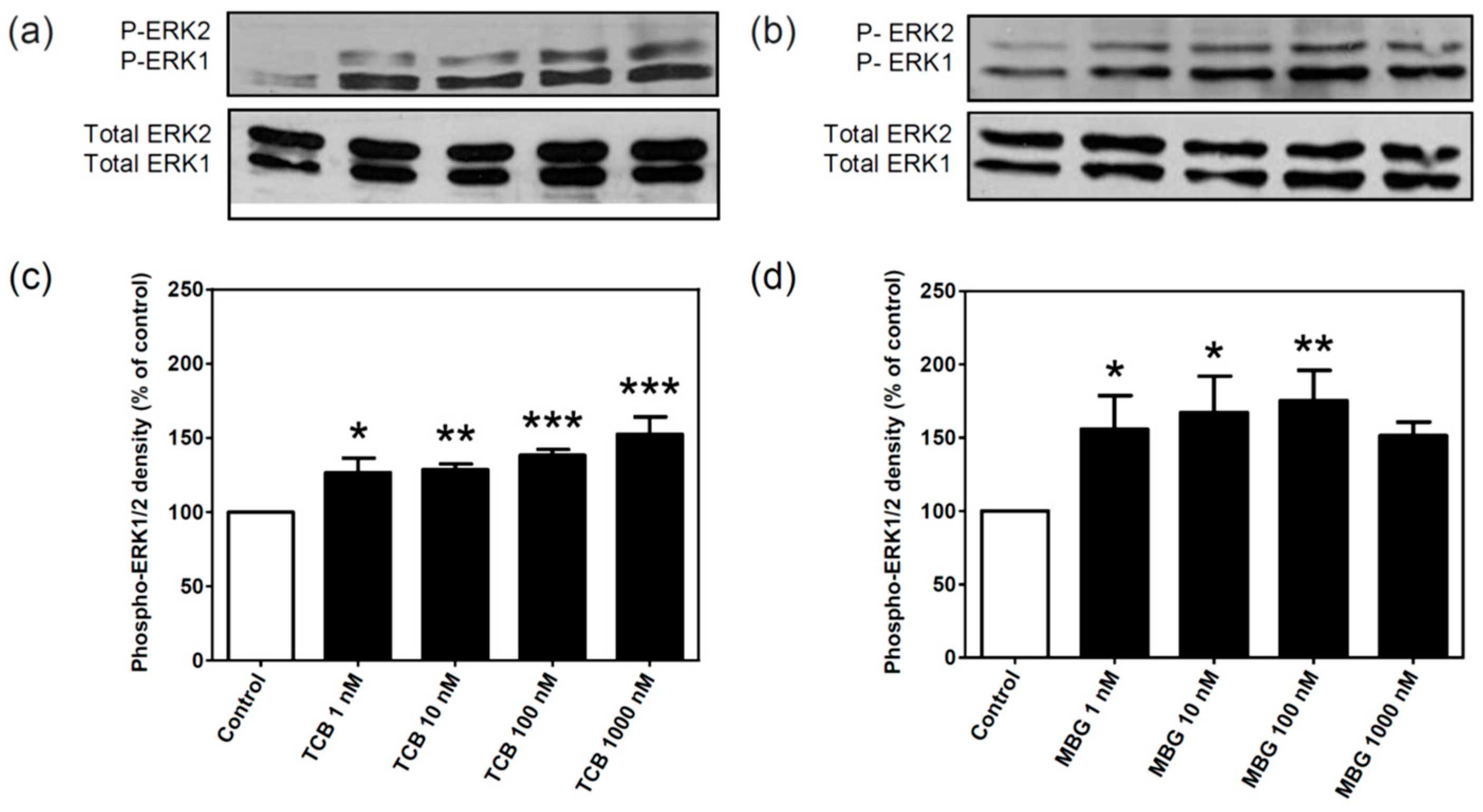
2.2. Effect of Bufadienolides on ERK1/2 Activation
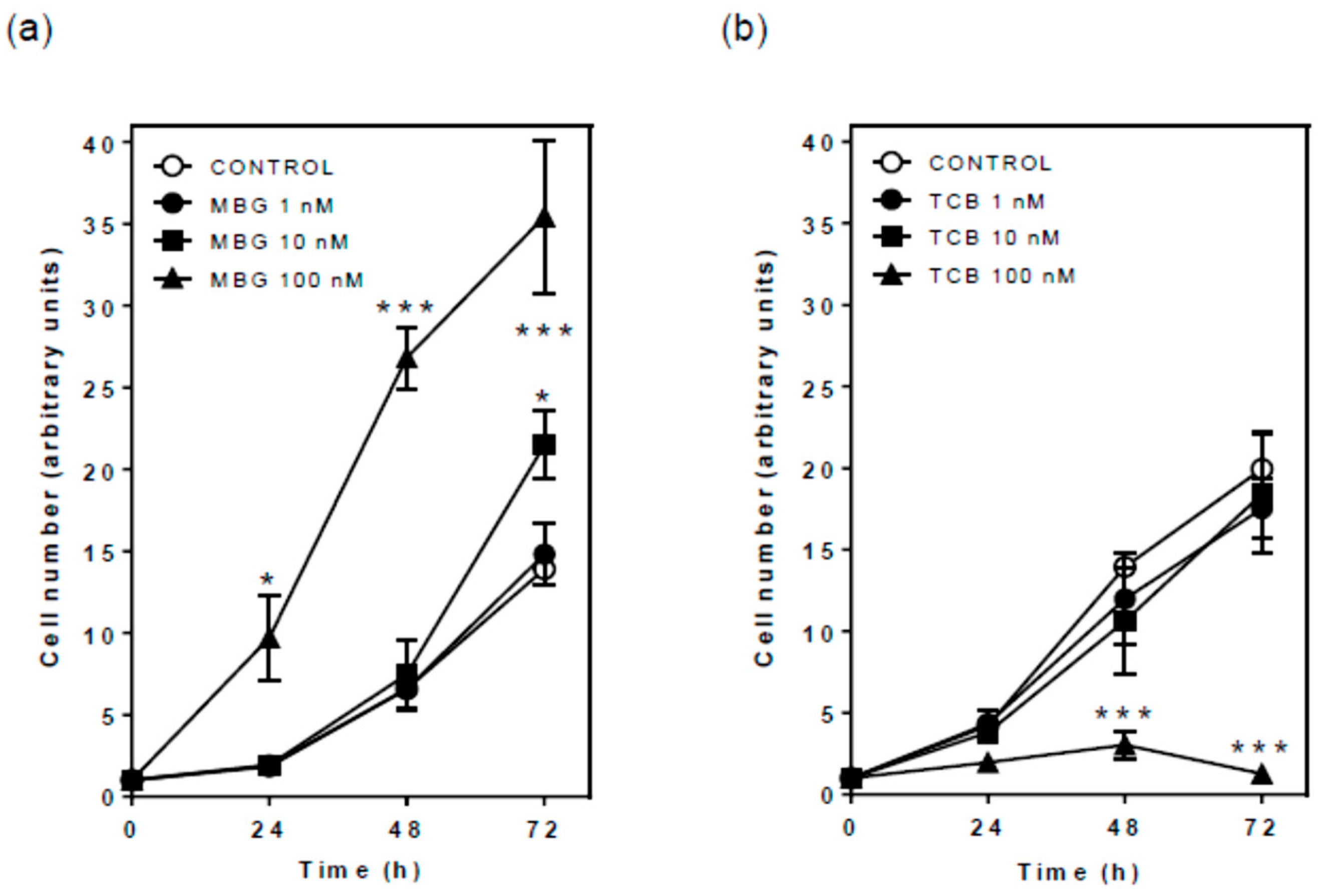
2.3. Effect of Bufadienolides on Cell Proliferation and Viability
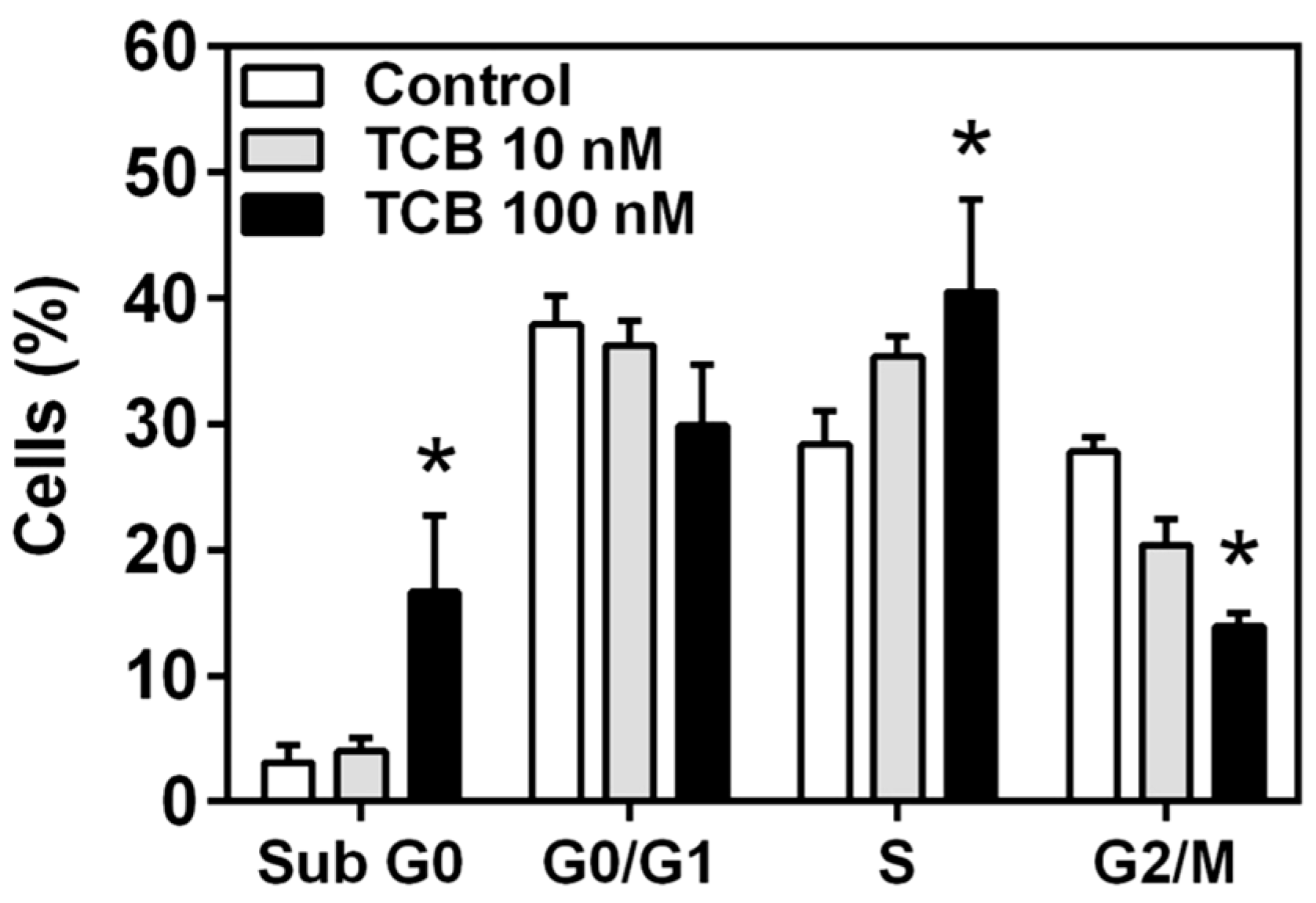
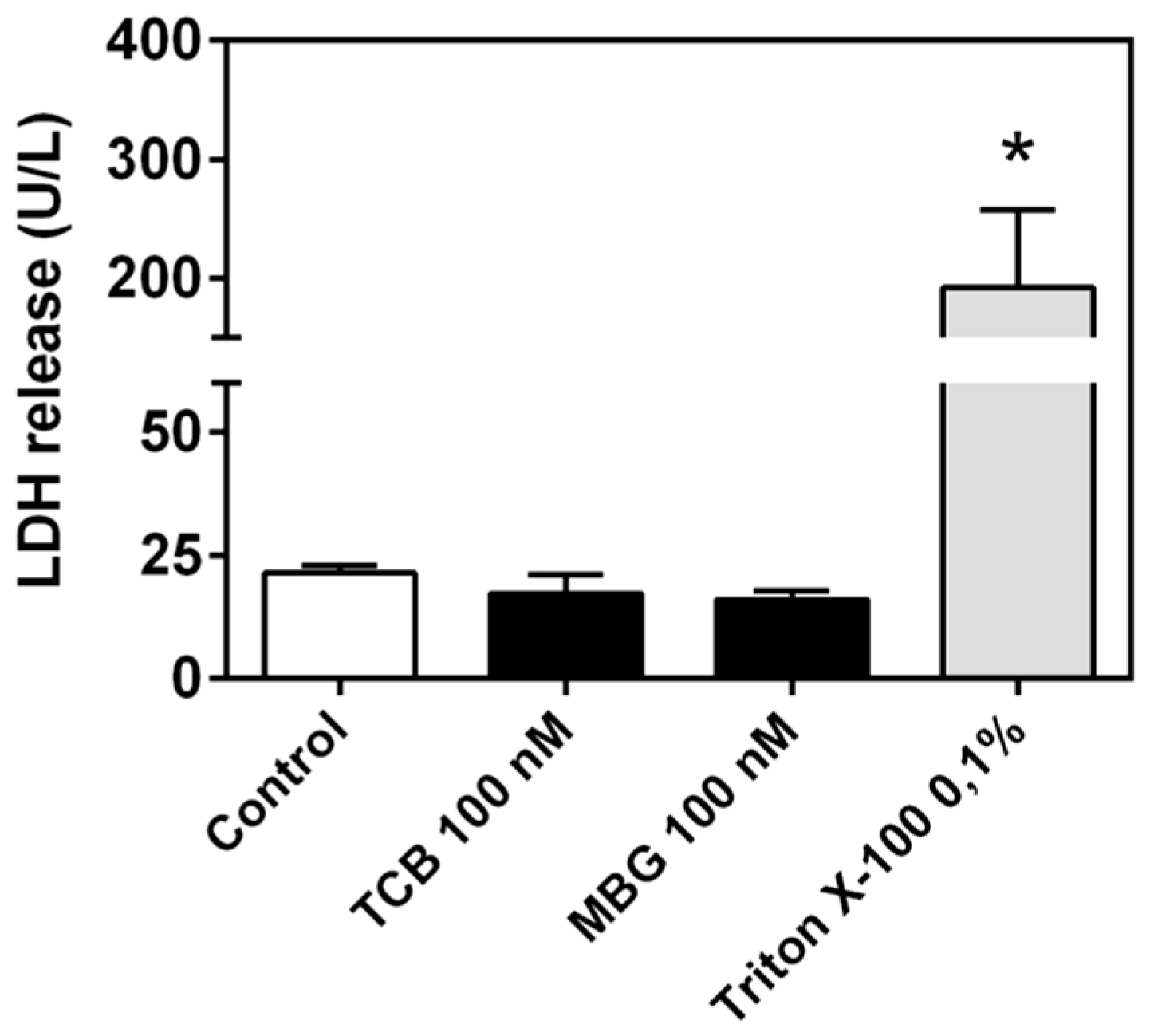
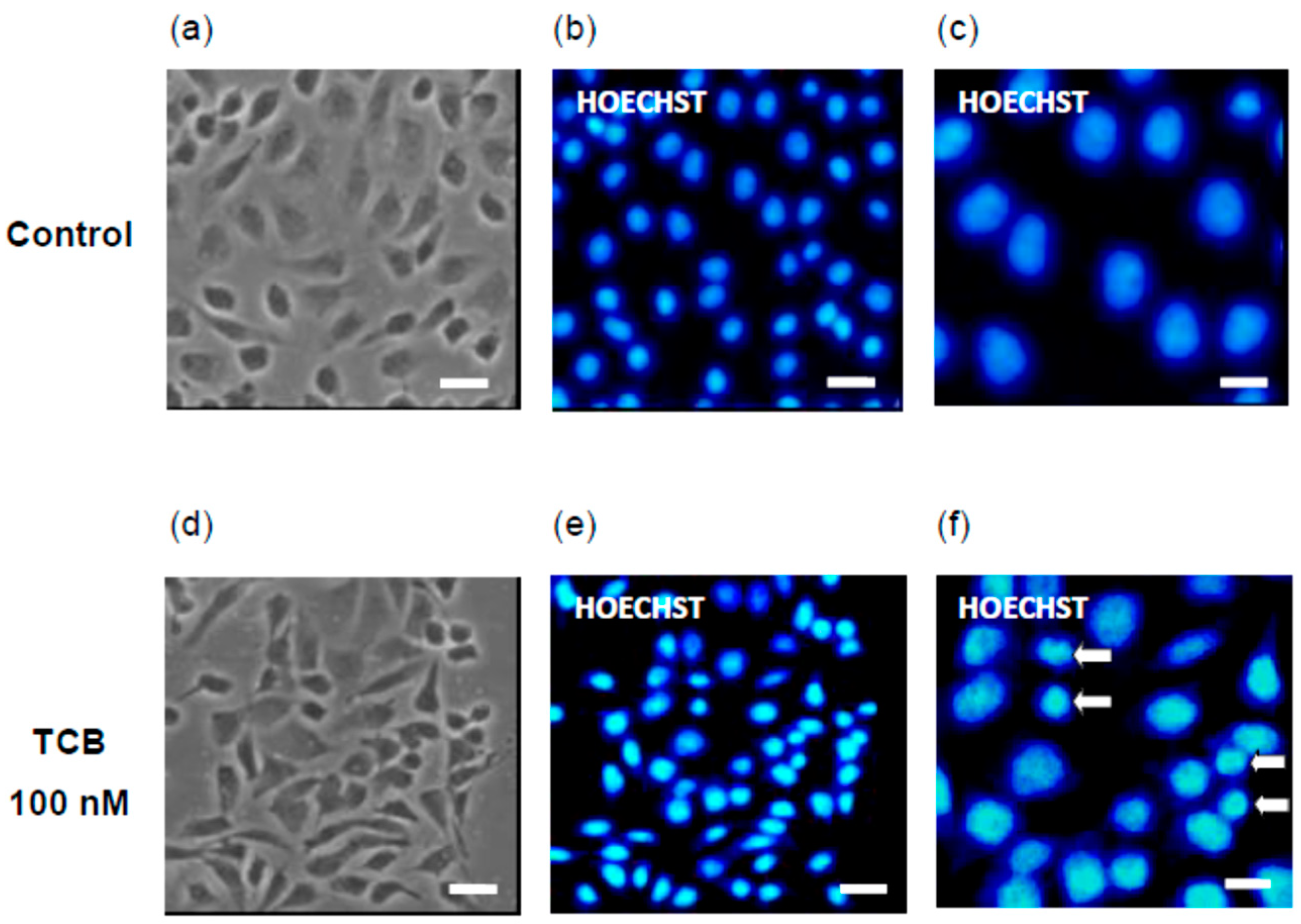
2.4. Effect of Telocinobufagin on Cell Cycle Phases and Cell Death
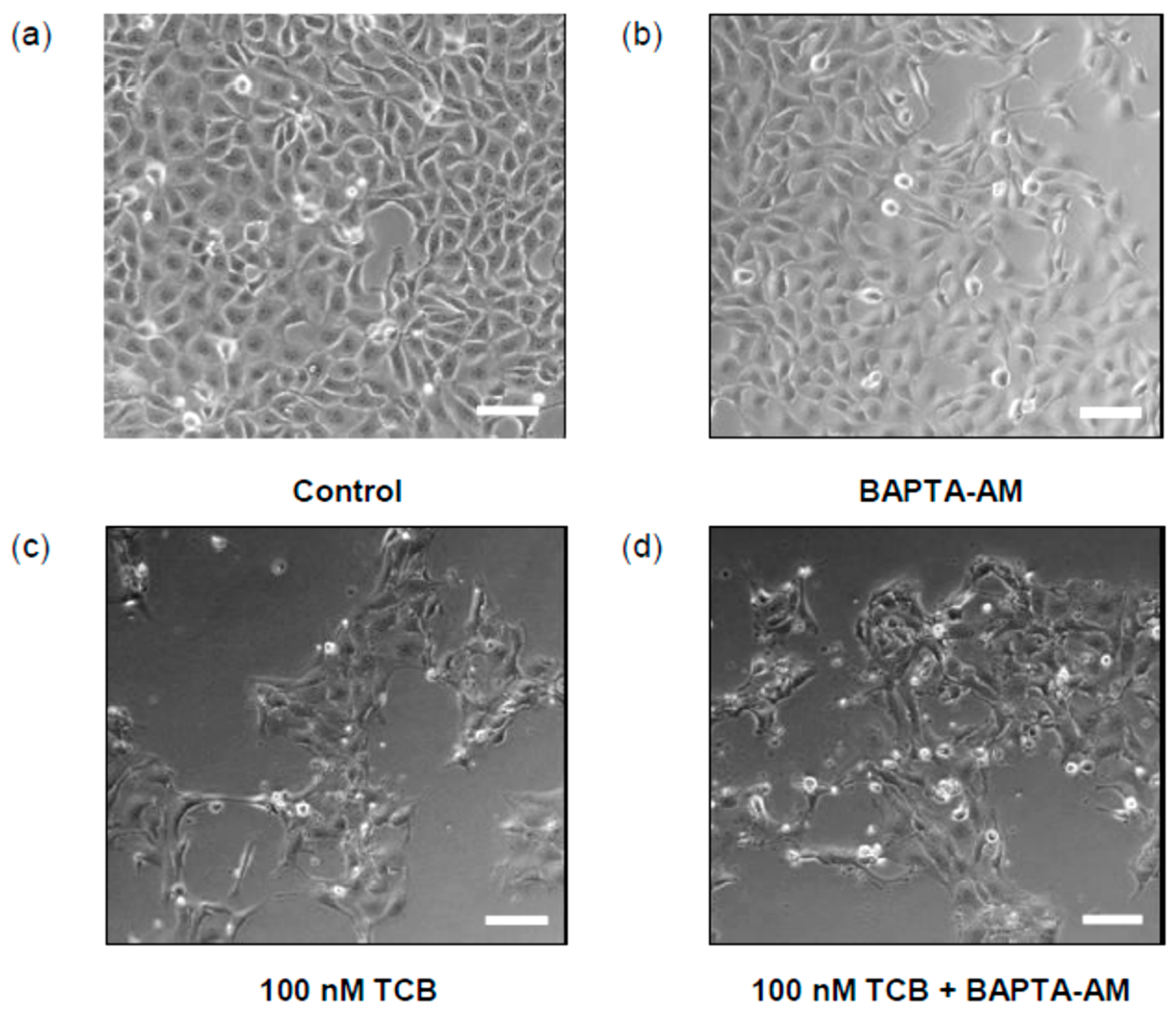
2.5. Role of Intracellular Ca2+ on Telocinobufagin-Induced Cell Death
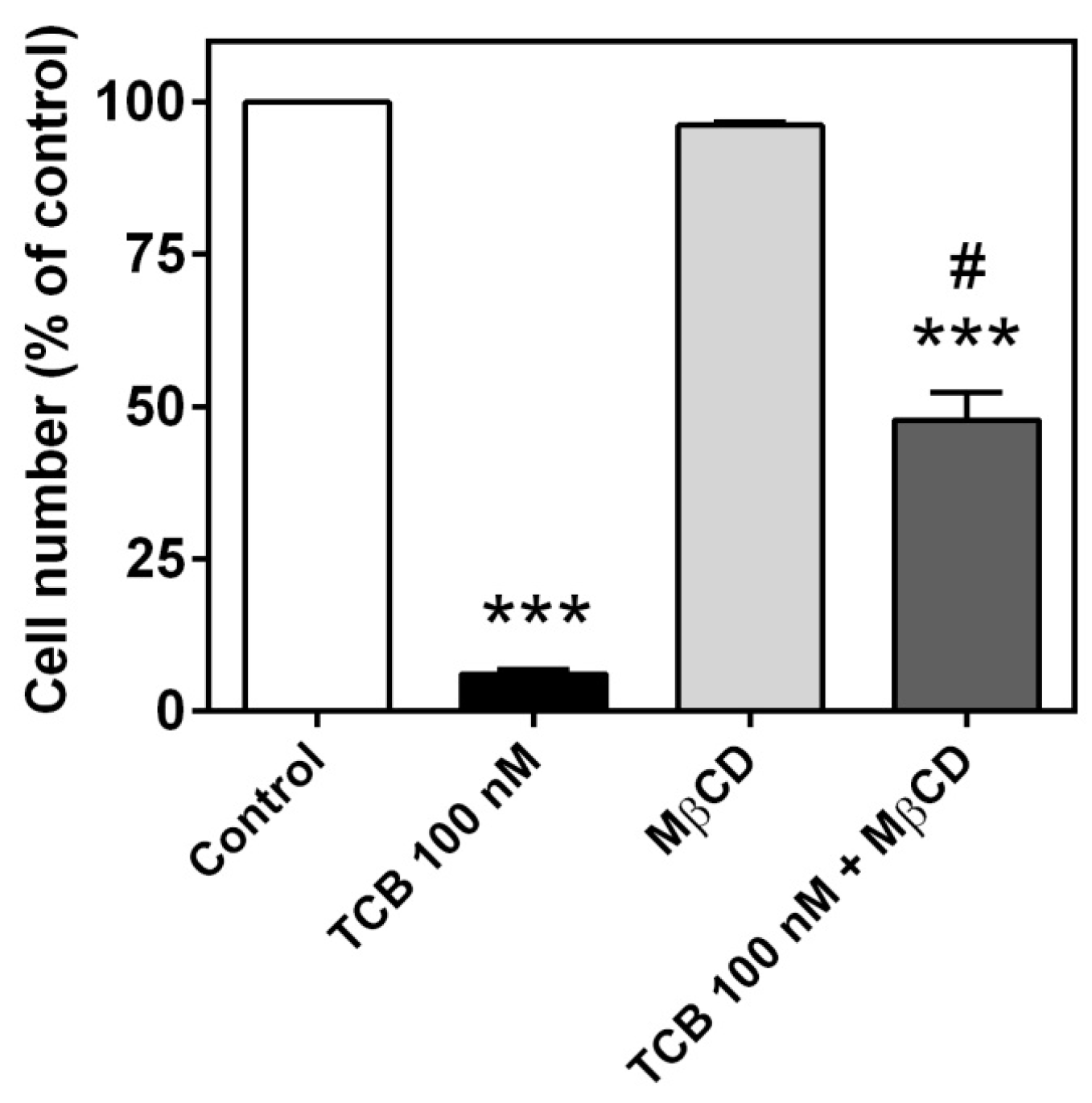
2.6. Role of Caveolae on Telocinobufagin-Induced Cell Death
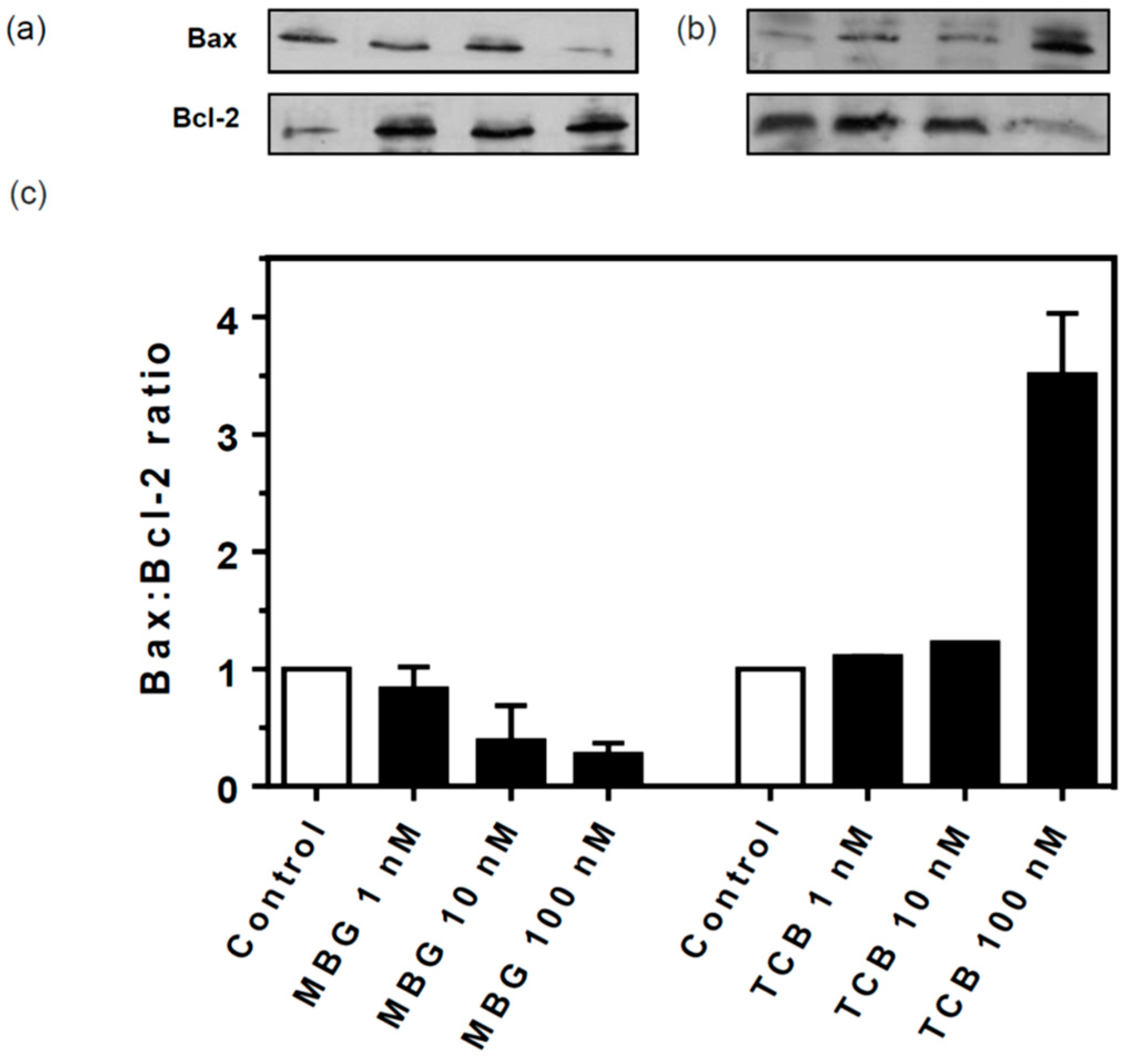
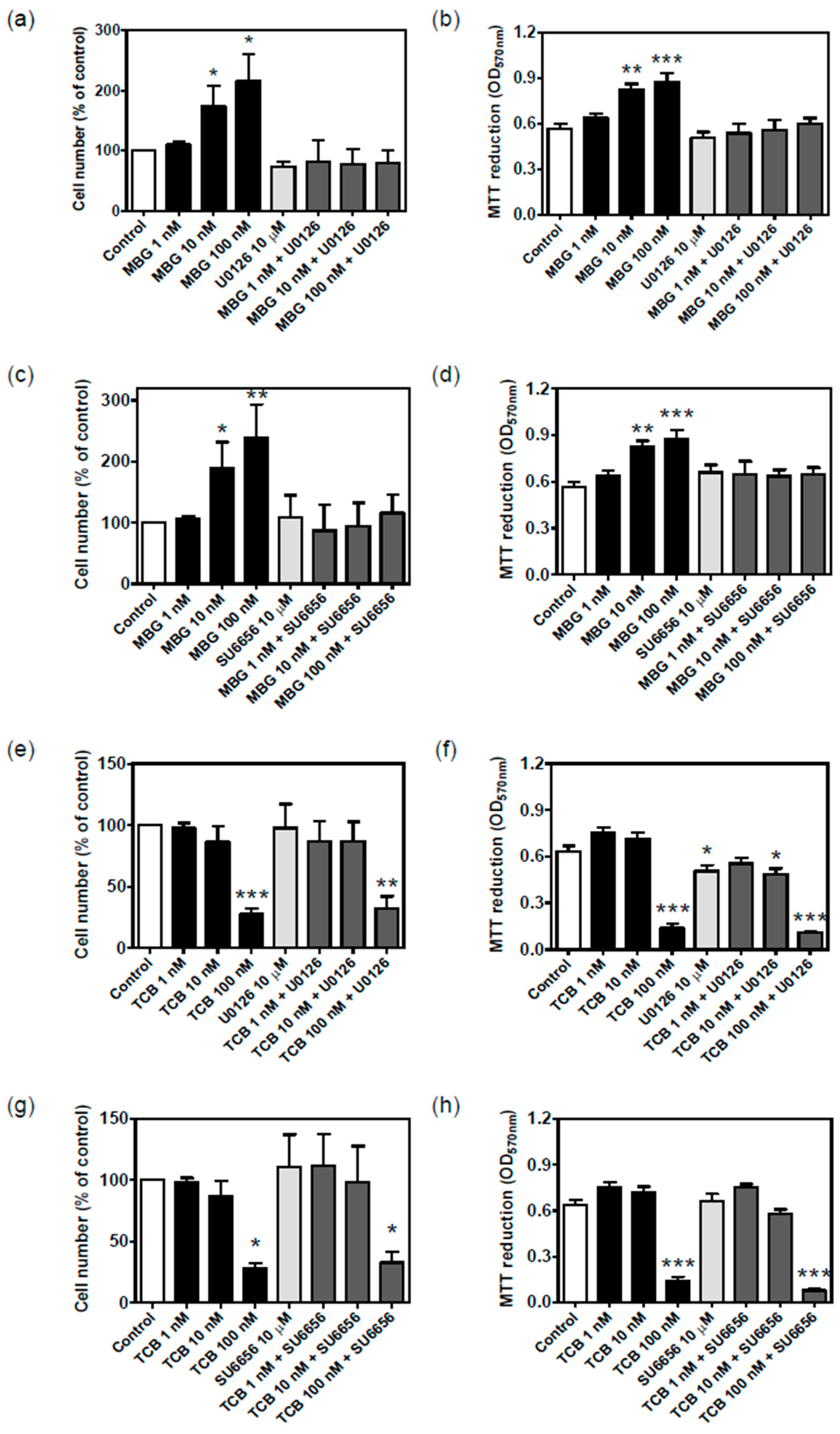
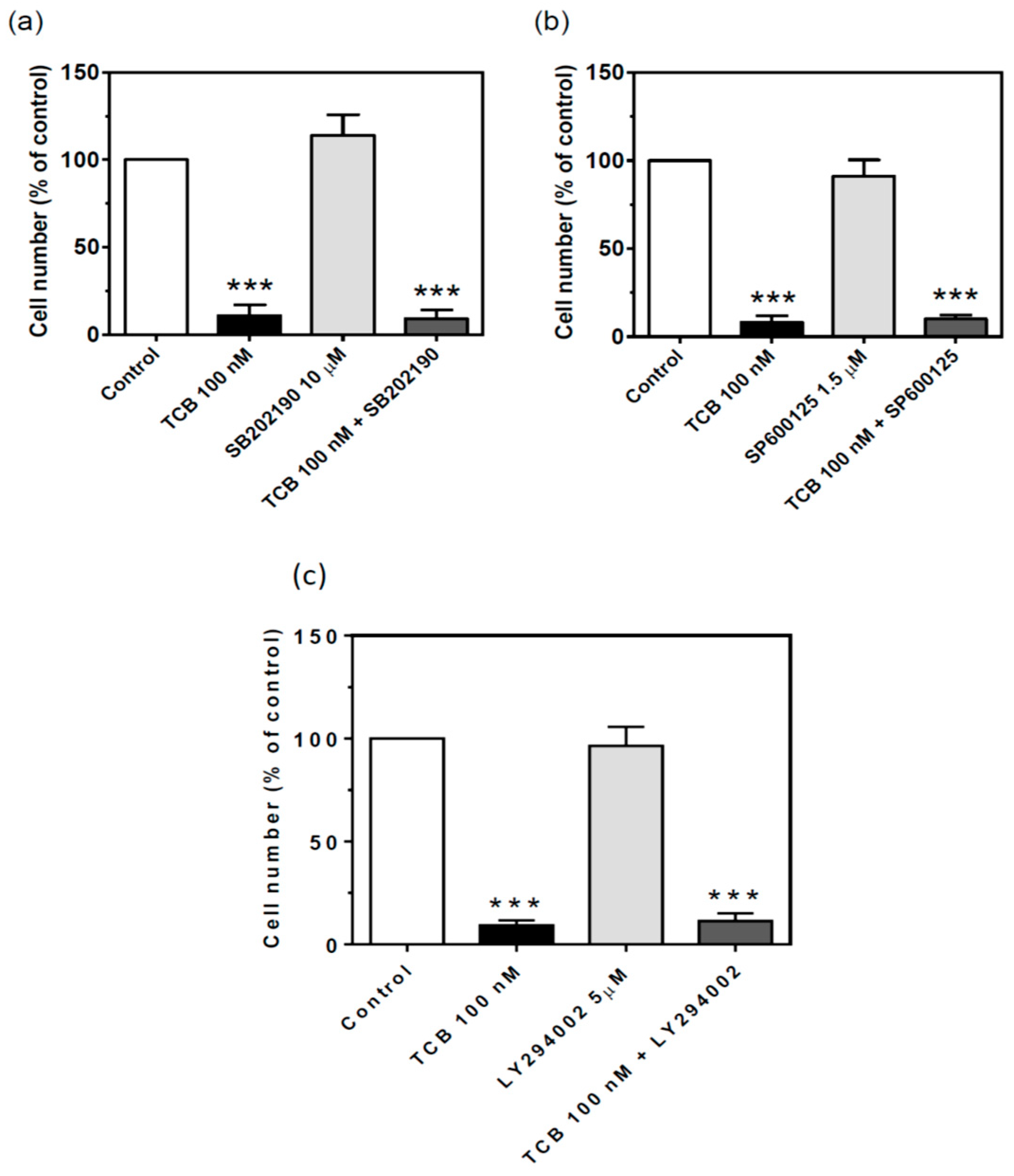
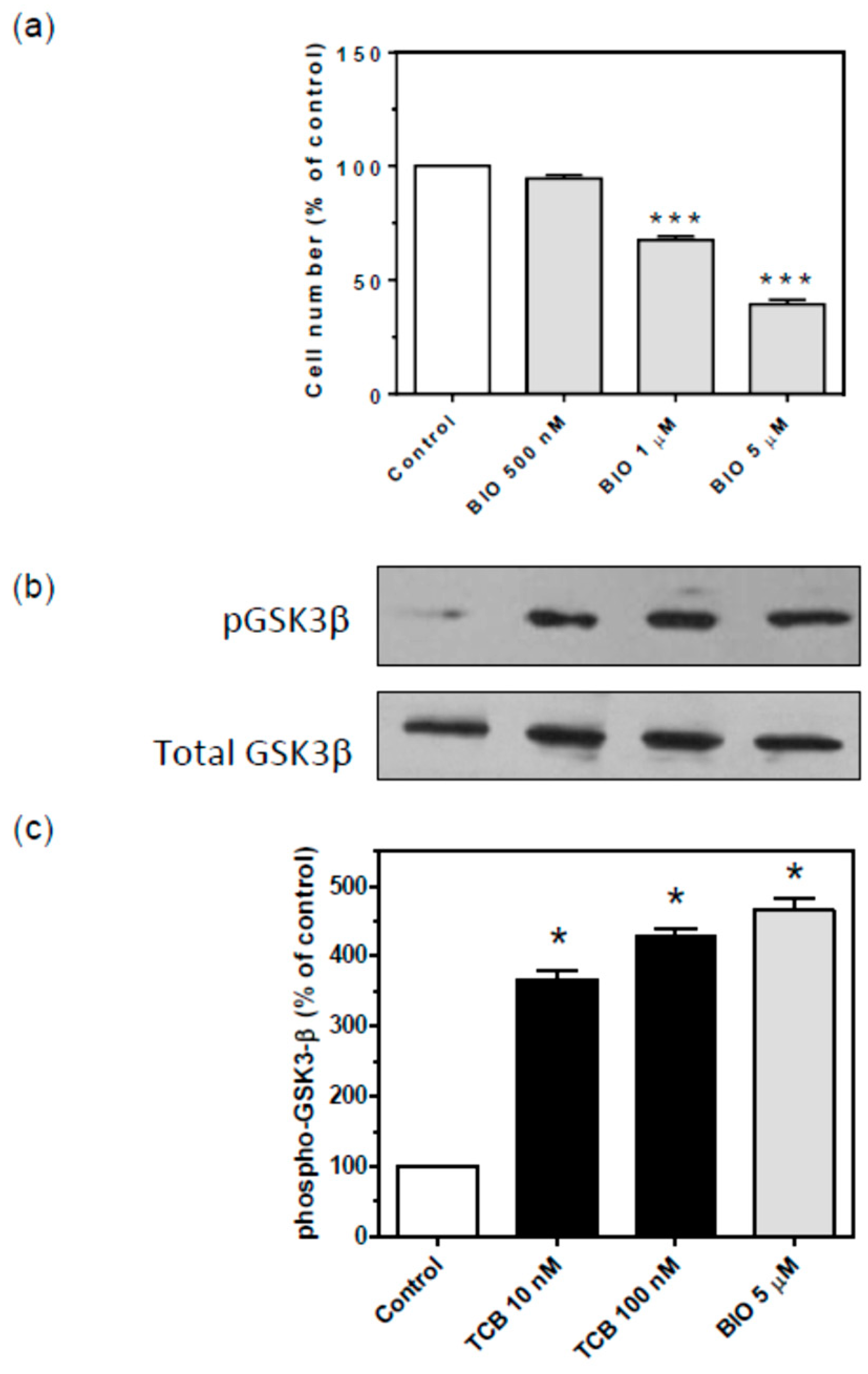
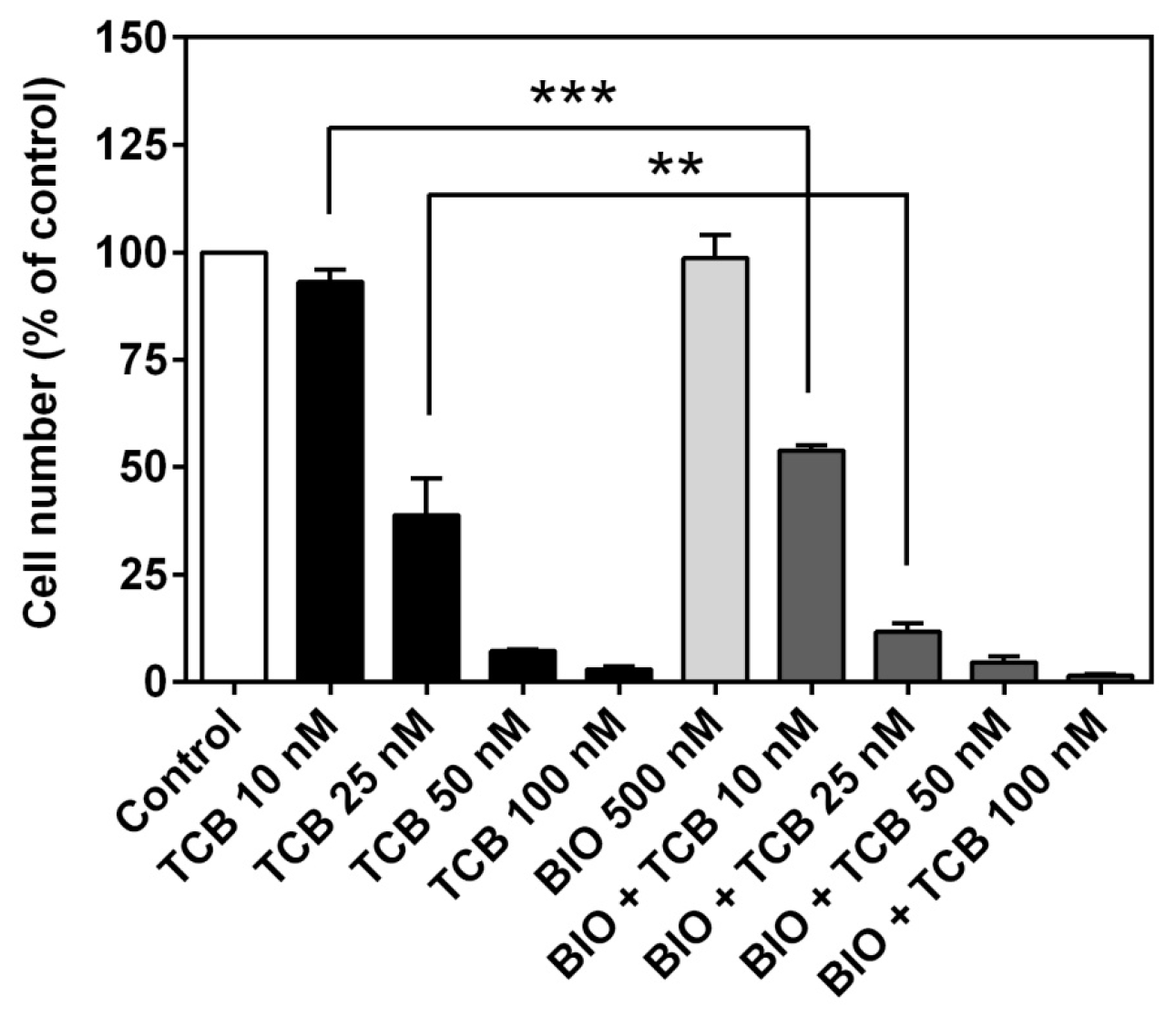
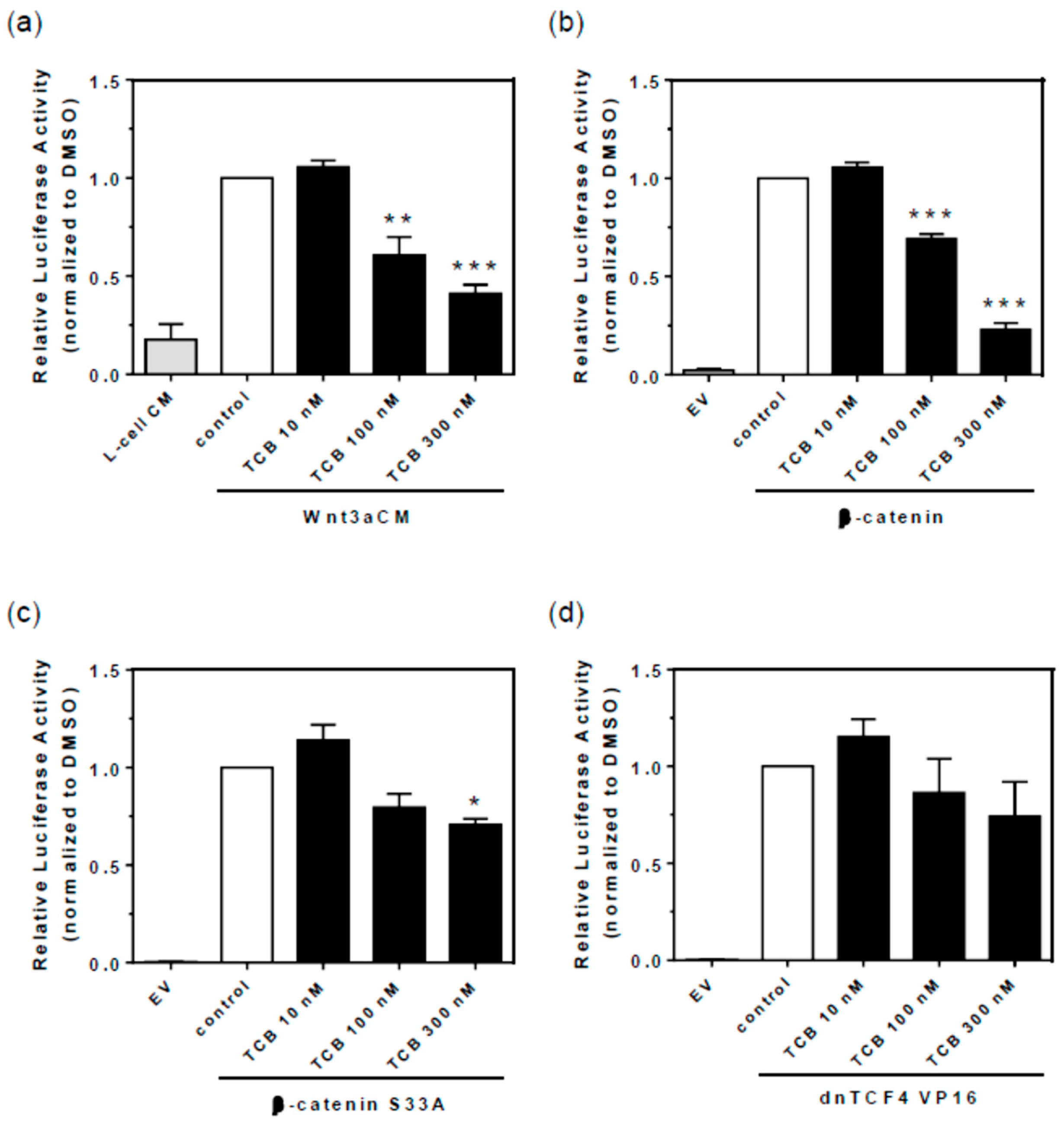
2.7. Signaling Pathways Involved in Bufadienolide Effect
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Culture
4.3. Cellular Preparation and Treatment with CTS
4.4. Na+/K+-ATPase Inhibition Assay
4.5. Immunoblot Assay
4.6. Cell Counting Assay
4.7. MTT Assay
4.8. LDH Release
4.9. Cell Cycle Analysis
4.10. Morphological Analysis by Phase-Contrast and Fluorescence Microscopy
4.11. Dual Luciferase Reporter Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Xie, Z.J. The Na,K-ATPase receptor complex: Its organization and membership. Cell Biochem. Biophys. 2006, 46, 303–316. [Google Scholar] [CrossRef]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Eur. J. Physiol. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Manunta, P.; Ferrari, P.; Bianchi, G. The endogenous ouabain: Molecular basis of its role in hypertension and cardiovascular complications. Front. Biosci. 2005, 10, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Nesher, M.; Shpolansky, U.; Rosen, H.; Lichtstein, D. The digitalis-like steroid hormones: New mechanisms of action and biological significance. Life Sci. 2007, 80, 2093–2107. [Google Scholar] [CrossRef] [PubMed]
- Puschett, J.B.; Agunanne, E.; Uddin, M.N. Emerging role of the bufadienolides in cardiovascular and kidney diseases. Am. J. Kidney Dis. 2010, 56, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.Q.; Machado, K.D.; Oliveira, S.F.; Araújo, L.D.; Monção-Filho, E.D.; Melo-Cavalcante, A.A.; Vieira-Júnior, G.M.; Ferreira, P.M. Bufadienolides from amphibians: A promising source of anticancer prototypes for radical innovation, apoptosis triggering and Na+/K+-ATPase inhibition. Toxicon 2017, 127, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 2007, 1776, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Cerella, C.; Guchelaar, H.J.; Diederich, M.; Gelderblom, H. Cardiac glycosides in cancer therapy: From preclinical investigations towards clinical trials. Investig. New Drugs 2013, 31, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-de-Albuquerque, C.F.; Ribeiro Silva, A.; Ignácio da Silva, C.; Castro-Faria-Neto, H.C.; Burth, P. Na/K-Pump and Beyond: Na/K-ATPase as a Modulator of Apoptosis and Autophagy. Molecules 2017, 22, E578. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Lin, Y.; Nutt, L.K.; Ozel, H.Z.; Newman, R.A. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000, 60, 3807–3812. [Google Scholar] [PubMed]
- Tian, J.; Li, X.; Liang, M.; Liu, L.; Xie, J.X.; Ye, Q.; Kometiani, P.; Tillekeratne, M.; Jin, R.; Xie, Z. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J. Biol. Chem. 2009, 284, 14921–14929. [Google Scholar] [CrossRef]
- Nesher, M.; Shpolansky, U.; Viola, N.; Dvela, M.; Buzaglo, N.; Cohen Ben-Ami, H.; Rosen, H.; Lichtstein, D. Ouabain attenuates cardiotoxicity induced by other cardiac steroids. Br. J. Pharmacol. 2010, 160, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manunta, P.; Hamilton, J.; Rogowski, A.C.; Hamilton, B.P.; Hamlyn, J.M. Chronic hypertension induced by ouabain but not digoxin in the rat: Antihypertensive effect of digoxin and digitoxin. Hypertens. Res. 2000, 23, S77–S85. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Linde, C.L.; Pulina, M.V.; Baryshnikov, S.G.; Papparella, I.; Hamlyn, J.M.; Golovina, V.A. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2013, 15, C324–C333. [Google Scholar] [CrossRef] [PubMed]
- Akimova, O.A.; Bagrov, A.Y.; Lopina, O.D.; Kamernitsky, A.V.; Tremblay, J.; Hamet, P.; Orlov, S.N. Cardiotonic steroids differentially affect intracellular Na+ and [Na+]i/[K+]i-independent signaling in C7-MDCK cells. J. Biol. Chem. 2005, 280, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Arnaud-Batista, F.J.; Costa, G.T.; Oliveira, I.M.; Costa, P.P.; Santos, C.F.; Fonteles, M.C.; Uchôa, D.E.; Silveira, E.R.; Cardi, B.A.; Carvalho, K.M.; et al. Natriuretic effect of bufalin in isolated rat kidneys involves activation of the Na+-K+-ATPase-Src kinase pathway. Am. J. Physiol. Renal Physiol. 2012, 302, F959–F966. [Google Scholar] [CrossRef] [PubMed]
- Galandrin, S.; Oligny-Longpré, G.; Bouvier, M. The evasive nature of drug efficacy: Implications for drug discovery. Trends Pharmacol. Sci. 2007, 28, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; Von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M.; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Quintas, L.E.M.; Pierre, S.V.; Liu, L.; Bai, Y.; Liu, X.; Xie, Z.J. Alterations of Na+/K+-ATPase function in caveolin-1 knockout cardiac fibroblasts. J. Mol. Cell. Cardiol. 2010, 49, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, J.; Dai, C.; Hirschi, K.K.; Dmitrieva, R.I.; Doris, P.A.; Liu, L.; Allen, J.C. Ouabain- and marinobufagenin-induced proliferation of human umbilical vein smooth muscle cells and rat vascular smooth muscle cell line, A7r5. Circulation 2003, 108, 3048–3053. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell. 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [PubMed]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mohammadi, K.; Aynafshar, B.; Wang, H.; Li, D.; Liu, J.; Ivanov, A.V.; Xie, Z.; Askari, A. Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am. J. Physiol. Cell Physiol. 2003, 284, C1550–C1560. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wu, J.; Li, D.; Morgan, E.E.; Liu, J.; Zhao, X.; Walsh, A.; Saikumar, J.; Tinkel, J.; Joe, B.; et al. Differential roles of caveolin-1 in ouabain-induced Na+/K+-ATPase cardiac signaling and contractility. Physiol. Genom. 2016, 48, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Haas, M.; Liang, M.; Cai, T.; Tian, J.; Li, S.; Xie, Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J. Biol. Chem. 2004, 279, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.N.; Jansson, K.; Sánchez, G.; Sharma, M.; Reif, G.A.; Wallace, D.P.; Blanco, G. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. Am. J. Physiol. Renal Physiol. 2011, 301, F897–F906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, T.N.; Possidonio, A.C.; Soares, C.P.; Ayres, R.; Costa, M.L.; Quintas, L.E.M.; Mermelstein, C. The role of Na+/K+-ATPase during chick skeletal myogenesis. PLoS ONE 2015, 10, e0120940. [Google Scholar] [CrossRef] [PubMed]
- Afroze, S.H.; Sloan, J.; Osuji, G.C.; Drever, N.; Pilkinton, K.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Cinobufotalin impedes Sw.71 cytotrophoblast cell line function via cell cycle arrest and apoptotic signaling. Mol. Cell. Biochem. 2016, 422, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Liu, L.; Askari, A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol. Pharmacol. 2005, 67, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.J.; Hu, L.P.; Peng, Q.L.; Yang, X.L.; Bai, L.L.; Yiu, A.; Li, Y.; Tian, H.Y.; Ye, W.C.; Zhang, D.M. Hellebrigenin induces cell cycle arrest and apoptosis in human hepatocellular carcinoma HepG2 cells through inhibition of Akt. Chem. Biol. Interact. 2014, 219, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, C.; Xu, L.; Zang, K.; Ning, Z.; Jiang, F.; Chi, H.; Zhu, X.; Meng, Z. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1α via the PI3K/AKT/mTOR pathway. Oncotarget 2016, 7, 20193–20208. [Google Scholar] [PubMed] [Green Version]
- Zhang, G.; Wang, C.; Sun, M.; Li, J.; Wang, B.; Jin, C.; Hua, P.; Song, G.; Zhang, Y.; Nguyen, L.L.; et al. Cinobufagin inhibits tumor growth by inducing intrinsic apoptosis through AKT signaling pathway in human nonsmall cell lung cancer cells. Oncotarget 2016, 7, 28935–28946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget 2017, 8, 14221–14250. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.H.; Liu, X.; Qiu, Y.Y.; Cai, J.F.; Qin, J.M.; Zhu, H.R.; Li, Q. Anti-tumor activity and apoptosis-regulation mechanisms of bufalin in various cancers: New hope for cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. Renal Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef]
- Gable, M.E.; Ellis, L.; Fedorova, O.V.; Bagrov, A.Y.; Askari, A. Comparison of digitalis sensitivities of Na+/K+-ATPases from human and pig kidneys. ACS Omega 2017, 2, 3610–3615. [Google Scholar] [CrossRef] [PubMed]
- Godinho, A.N.; Costa, G.T.; Oliveira, N.O.; Cardi, B.A.; Uchoa, D.E.A.; Silveira, E.R.; Quintas, L.E.M.; Noël, F.; Fonteles, M.C.; Carvalho, K.M.; et al. Effects of cardiotonic steroids on isolated perfused kidney and NHE3 activity in renal proximal tubules. Biochim. Biophys. Acta 2017, 1861, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Touza, N.A.; Pôças, E.S.; Quintas, L.E.M.; Cunha-Filho, G.; Santos, M.L.; Noël, F. Inhibitory effect of combinations of digoxin and endogenous cardiotonic steroids on Na+/K+-ATPase activity in human kidney membrane preparation. Life Sci. 2011, 88, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Cai, T. Na+-K+-ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules 2017, 22, E990. [Google Scholar] [PubMed]
- Trevisi, L.; Visentin, B.; Cusinato, F.; Pighin, I.; Luciani, S. Antiapoptotic effect of ouabain on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 321, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Khundmiri, S.J.; Amin, V.; Henson, J.; Lewis, J.; Ameen, M.; Rane, M.J.; Delamere, N.A. Ouabain stimulates protein kinase B (Akt) phosphorylation in opossum kidney proximal tubule cells through an ERK-dependent pathway. Am. J. Physiol. Cell Physiol. 2007, 293, C1171–C1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.N.; Wallace, D.P.; Blanco, G. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J. Am. Soc. Nephrol. 2007, 18, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Soares-da-Silva, P. Long-term regulation of Na+,K+-ATPase in opossum kidney cells by ouabain. J. Cell Physiol. 2011, 226, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.F.; Amaral, L.S.; Porto, C.S.; Quintas, L.E.M. Na+/K+-ATPase α1 isoform mediates ouabain-induced expression of cyclin D1 and proliferation of rat sertoli cells. Reproduction 2012, 144, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [PubMed]
- Sayed, M.; Drummond, C.A.; Evans, K.L.; Haller, S.T.; Liu, J.; Xie, Z.; Tian, J. Effects of Na/K-ATPase and its ligands on bone marrow stromal cell differentiation. Stem Cell Res. 2014, 13, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitrieva, R.I.; Doris, P.A. Ouabain is a potent promoter of growth and activator of ERK1/2 in ouabain-resistant rat renal epithelial cells. J. Biol. Chem. 2003, 278, 28160–28166. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.; Eva, A.; Kirch, U.; Boldyrev, A.; Scheiner-Bobis, G. Ouabain activates signaling pathways associated with cell death in human neuroblastoma. Biochim. Biophys. Acta 2007, 1768, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Karpova, L.V.; Bulygina, E.R.; Boldyrev, A.A. Different neuronal Na+/K+-ATPase isoforms are involved in diverse signaling pathways. Cell Biochem. Funct. 2010, 28, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Chueh, S.; Guh, J.; Chen, J.; Lai, M.; Teng, C. Dual effects of ouabain on the regulation of proliferation and apoptosis in human prostatic smooth muscle cells. J. Urol. 2001, 66, 347–353. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Guan, L. Effects of ouabain on proliferation intracellular free calcium and c-myc mRNA expression in vascular smooth muscle cells. J. Comp. Physiol. B 2007, 177, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Olej, B.; dos Santos, N.F.; Leal, L.; Rumjanek, V.M. Ouabain induces apoptosis on PHA-activated lymphocytes. Biosci. Rep. 1998, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Taurin, S.; Der Sarkissian, S.; Lopina, O.D.; Pshezhetsky, A.V.; Tremblay, J.; deBlois, D.; Hamet, P.; Orlov, S.N. Inhibition of Na+,K+-ATPase by ouabain triggers epithelial cell death independently of inversion of the [Na+]i/[K+]i ratio. Biochem. Biophys. Res. Commun. 2003, 301, 735–744. [Google Scholar] [CrossRef]
- Orlov, S.N.; Thorin-Trescases, N.; Pchejetski, D.; Taurin, S.; Farhat, N.; Tremblay, J.; Thorin, E.; Hamet, P. Na+/K+ pump and endothelial cell survival: [Na+]i/[K+]i-independent necrosis triggered by ouabain, and protection against apoptosis mediated by elevation of [Na+]i. Pflugers Arch. 2004, 448, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Winnicka, K.; Bielawski, K.; Bielawska, A.; Miltyk, W. Dual effects of ouabain, digoxin and proscillaridin A on the regulation of apoptosis in human fibroblasts. Nat. Prod. Res. 2010, 24, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Cardiotonic steroids-mediated targeting of the Na+/K+-ATPase to combat chemoresistant cancers. Curr. Med. Chem. 2012, 19, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Akera, T.; Brody, T.M. The role of Na+,K+-ATPase in the inotropic action of digitalis. Pharmacol. Rev. 1978, 29, 187–220. [Google Scholar]
- Lin, H.; Juang, J.L.; Wang, P.S. Involvement of Cdk5/p25 in digoxin-triggered prostate cancer cell apoptosis. J. Biol. Chem. 2004, 279, 29302–29307. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.Y.; Huang, W.J.; Kan, S.F.; Wang, P.S. Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003, 54, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wu, L.; Qu, W.; Malhotra, D.; Xie, Z.; Shapiro, J.I.; Liu, J. Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 2008, 294, C555–C563. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Dicato, M.; Diederich, M. Assembling the puzzle of anti-cancer mechanisms triggered by cardiac glycosides. Mitochondrion 2013, 13, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Akimova, O.A.; Lopina, O.D.; Rubtsov, A.M.; Gekle, M.; Tremblay, J.; Hamet, P.; Orlov, S.N. Death of ouabain-treated renal epithelial cells: Evidence for p38 MAPK-mediated Nai+/Ki+-independent signaling. Apoptosis 2009, 14, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Akimova, O.A.; Lopina, O.D.; Rubtsov, A.M.; Hamet, P.; Orlov, S.N. Investigation of mechanism of p38 MAPK activation in renal epithelial cell from distal tubules triggered by cardiotonic steroids. Biochemistry 2010, 75, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Ehrig, J.C.; Afroze, S.H.; Reyes, M.; Allen, S.R.; Drever, N.S.; Pilkinton, K.A.; Kuehl, T.J.; Uddin, M.N. A p38 mitogen-activated protein kinase inhibitor attenuates cardiotonic steroids-induced apoptotic and stress signaling in a Sw-71 cytotrophoblast cell line. Placenta 2015, 36, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Grumati, P.; Cusinato, F.; Orso, G.; Bonaldo, P.; Trevisi, L. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem. Pharmacol. 2014, 89, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Q.; Pang, J.; Jin, H.; Li, H.; Yang, X. Blocking autophagy enhances the pro-apoptotic effect of bufalin on human gastric cancer cells through endoplasmic reticulum stress. Biol. Open 2017, 6, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotova, O.; Al-Khalili, L.; Talia, S.; Hooke, C.; Fedorova, O.V.; Bagrov, A.Y.; Chibalin, A.V. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J. Biol. Chem. 2006, 281, 20085–20094. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.Q.; Sheng, X.; Qin, J.M.; Sun, K.; Zhao, W.; Ni, L. The effect and mechanism of bufalin on regulating hepatocellular carcinoma cell invasion and metastasis via Wnt/β-catenin signaling pathway. Int. J. Oncol. 2016, 48, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Sowa, Y.; Iizumi, Y.; Aono, Y.; Sakai, T. Resibufogenin induces G1-phase arrest through the proteasomal degradation of cyclin D1 in human malignant tumor cells. PLoS ONE 2015, 10, e0129851. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.H.; Zhang, J.H.; Zhang, Q.Q.; Cui, Y.H.; Wang, Y.; Kou, W.Z.; Miao, Z.H.; Lu, P.; Wang, L.F.; Xu, Z.Y.; et al. Degradation of Mcl-1 through GSK-3β activation regulates apoptosis induced by bufalin in non-small cell lung cancer H1975 cells. Cell Physiol. Biochem. 2017, 41, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.K.; Bouchie, M.P.; Kukuruzinska, M.A. N-Glycosylation gene DPAGT1 is a target of the Wnt/β-catenin signaling pathway. J. Biol. Chem. 2010, 285, 31164–31173. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Awada, C.; Matsumoto, S.; Kaneiwa, T.; Sugimoto, T.; Takao, T.; Kikuchi, A. Basolateral secretion of Wnt5a in polarized epithelial cells is required for apical lumen formation. J. Cell Sci. 2015, 128, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhu, B.; Liu, X.; Yu, H.; Yong, L.; Liu, X.; Shao, J.; Liu, Z. Inhibition of oleandrin on the proliferation show and invasion of osteosarcoma cells in vitro by suppressing Wnt/β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 2015, 34, 115. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, Q.; Tian, Y. Reversal effect of ouabain on multidrug resistance in esophageal carcinoma EC109/CDDP cells by inhibiting the translocation of Wnt/β-catenin into the nucleus. Tumour Biol. 2016, 37, 15937–15947. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, L.V.; Raju, V.; El-Okdi, N.; Shidyak, A.; Kennedy, D.J.; Vetteth, S.; Giovannucci, D.R.; Bagrov, A.Y.; Fedorova, O.V.; Shapiro, J.I.; et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: Implication of epithelial-to-mesenchymal transition. Am. J. Physiol. Renal Physiol. 2009, 296, F922–F934. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, J.; McDermott, J.; Sanchez, G.; Sharma, M.; Barbosa, L.; Reif, G.A.; Wallace, D.P.; Blanco, G. Ouabain promotes partial epithelial to mesenchymal transition (EMT) changes in human autosomal dominant polycystic kidney disease (ADPKD) cells. Exp. Cell Res. 2017, 355, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Larre, I.; Ponce, A.; Fiorentino, R.; Shoshani, L.; Contreras, R.G.; Cereijido, M. Contacts and cooperation between cells depend on the hormone ouabain. Proc. Natl. Acad. Sci. USA 2006, 103, 10911–10916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T.; Williams, M. Defining and characterizing drug/compound function. Biochem. Pharmacol. 2014, 87, 40–63. [Google Scholar] [CrossRef] [PubMed]
- Runge, T.M.; Stephens, J.C.; Holden, P.; Havemann, D.F.; Kilgore, W.M.; Dale, E.M.; Dalton, R.E. Pharmacodynamic distinctions between ouabain, digoxin and digitoxin. Arch. Int. Pharmacodyn. Ther. 1975, 214, 31–45. [Google Scholar] [PubMed]
- Joubert, P.H. Are all cardiac glycosides pharmacodynamically similar? Eur. J. Clin. Pharmacol. 1990, 39, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Pamnani, M.B.; Chen, S.; Bryant, H.J.; Schooley, J.F., Jr.; Eliades, D.C.; Yuan, C.M.; Haddy, F.J. Effects of three sodium-potassium adenosine triphosphatase inhibitors. Hypertension 1991, 18, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.; Gregersen, J.L.; Yatime, L.; Nissen, P.; Fedosova, N.U. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. USA 2015, 112, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Klimanova, E.A.; Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Orlov, S.N.; Makarov, A.A.; Lopina, O.D. Binding of ouabain and marinobufagenin leads to different structural changes in Na,K-ATPase and depends on the enzyme conformation. FEBS Lett. 2015, 589, 2668–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Karashima, E.; Hamlyn, J.M.; Blaustein, M.P. Ouabain-digoxin antagonism in rat arteries and neurones. J. Physiol. 2014, 592, 941–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, T.; Glukmann, V.; Medvenev, E.; Shpolansky, U.; Galili, D.; Lichtstein, D.; Rosen, H. Role of endosomal Na+-K+-ATPase and cardiac steroids in the regulation of endocytosis. Am. J. Physiol. Cell Physiol. 2007, 293, C885–C896. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Filho, G.A.; Resck, I.S.; Cavalcanti, B.C.; Pessoa, C.O.; Moraes, M.O.; Ferreira, J.R.; Rodrigues, F.A.; Dos Santos, M.L. Cytotoxic profile of natural and some modified bufadienolides from toad Rhinella schneideri parotoid gland secretion. Toxicon 2010, 56, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Sottejeau, Y.; Gourbeau, J.M.; Sánchez, G.; Shidyak, A.; Blanco, G. Isoform specificity of Na-K-ATPase-mediated ouabain signaling. Am. J. Physiol. Renal Physiol. 2008, 294, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.L. Techniques for the study of steroid effects on membraneous (Na+ + K+)-ATPase. Methods Enzymol. 1975, 36, 434–439. [Google Scholar] [PubMed]
- Fontes, C.F.; Lopes, F.E.; Scofano, H.M.; Barrabin, H.; Norby, J.G. Stimulation of ouabain binding to Na,K-ATPase in 40% dimethyl sulfoxide by a factor from Na,K-ATPase preparations. Arch. Biochem. Biophys. 1999, 366, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Fiske, C.H.; Subbarow, Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar]














© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral, L.S.; Martins Ferreira, J.; Predes, D.; Abreu, J.G.; Noël, F.; Quintas, L.E.M. Telocinobufagin and Marinobufagin Produce Different Effects in LLC-PK1 Cells: A Case of Functional Selectivity of Bufadienolides. Int. J. Mol. Sci. 2018, 19, 2769. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092769
Amaral LS, Martins Ferreira J, Predes D, Abreu JG, Noël F, Quintas LEM. Telocinobufagin and Marinobufagin Produce Different Effects in LLC-PK1 Cells: A Case of Functional Selectivity of Bufadienolides. International Journal of Molecular Sciences. 2018; 19(9):2769. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092769
Chicago/Turabian StyleAmaral, Luciana S., Jainne Martins Ferreira, Danilo Predes, José Garcia Abreu, François Noël, and Luis Eduardo M. Quintas. 2018. "Telocinobufagin and Marinobufagin Produce Different Effects in LLC-PK1 Cells: A Case of Functional Selectivity of Bufadienolides" International Journal of Molecular Sciences 19, no. 9: 2769. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092769