Therapeutic Peptide Amphiphile as a Drug Carrier with ATP-Triggered Release for Synergistic Effect, Improved Therapeutic Index, and Penetration of 3D Cancer Cell Spheroids


Abstract
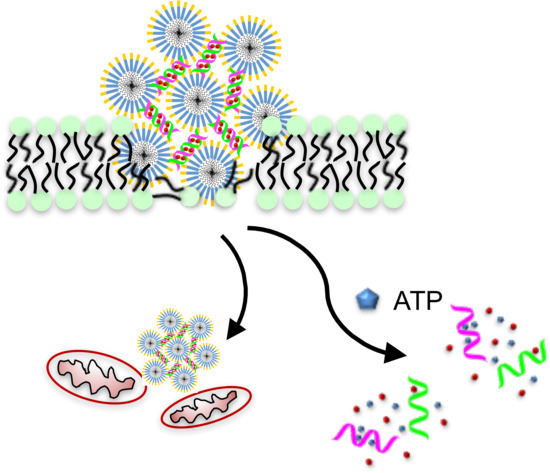
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
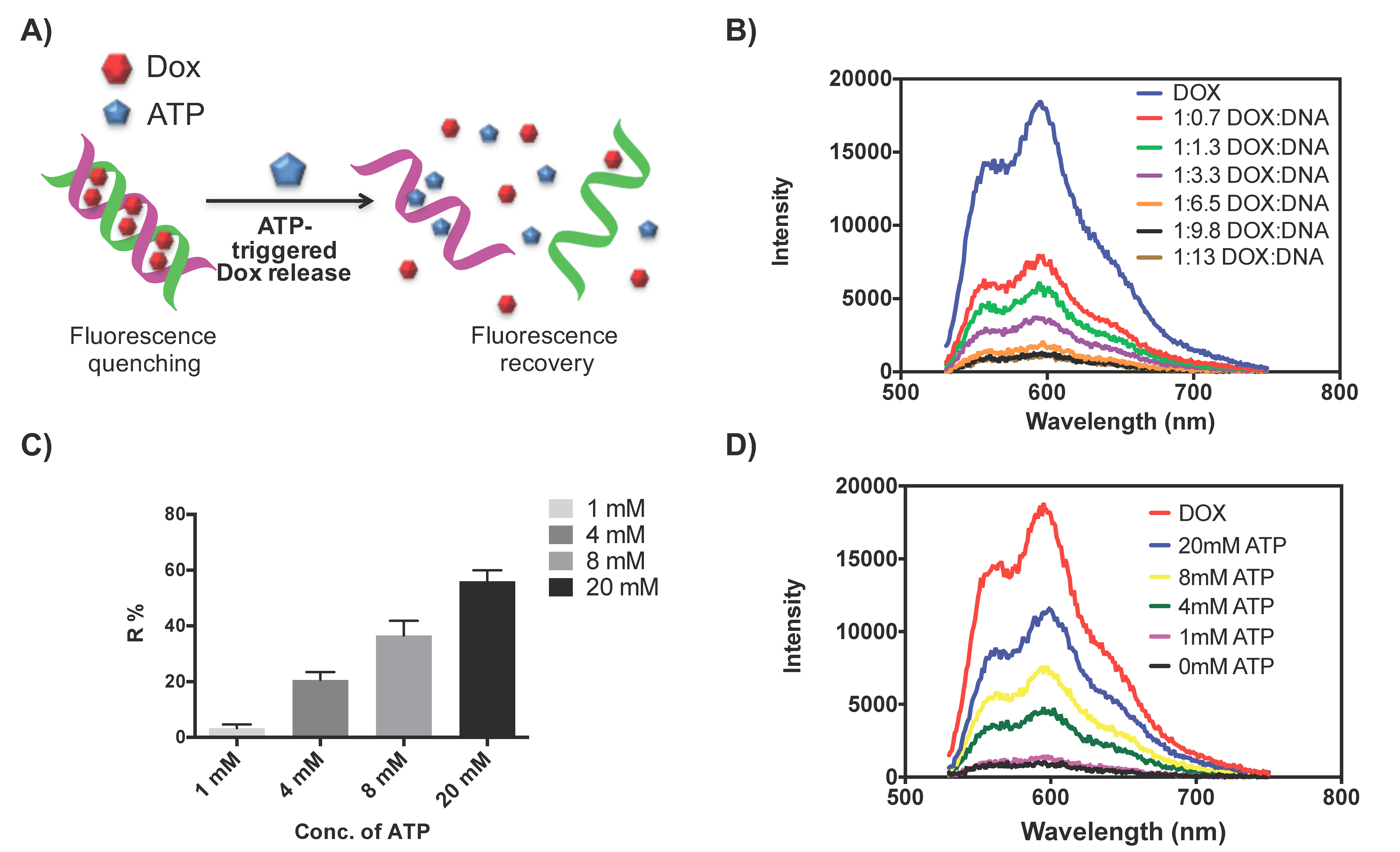
2.1. Dox Loading and Release from DNA Scaffold
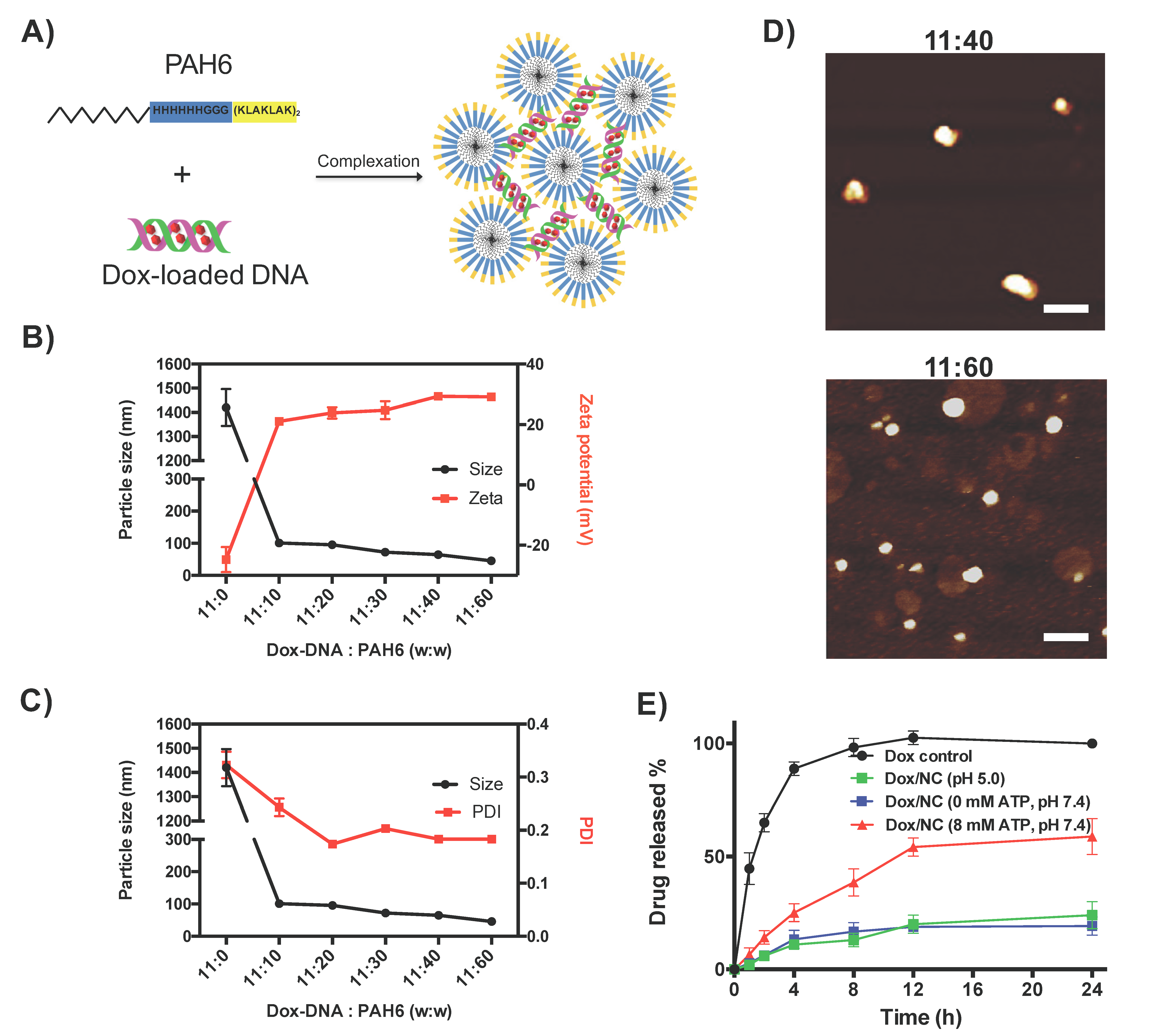
2.2. Characterization of the Dox-DNA/PAH6 Nanocomplex
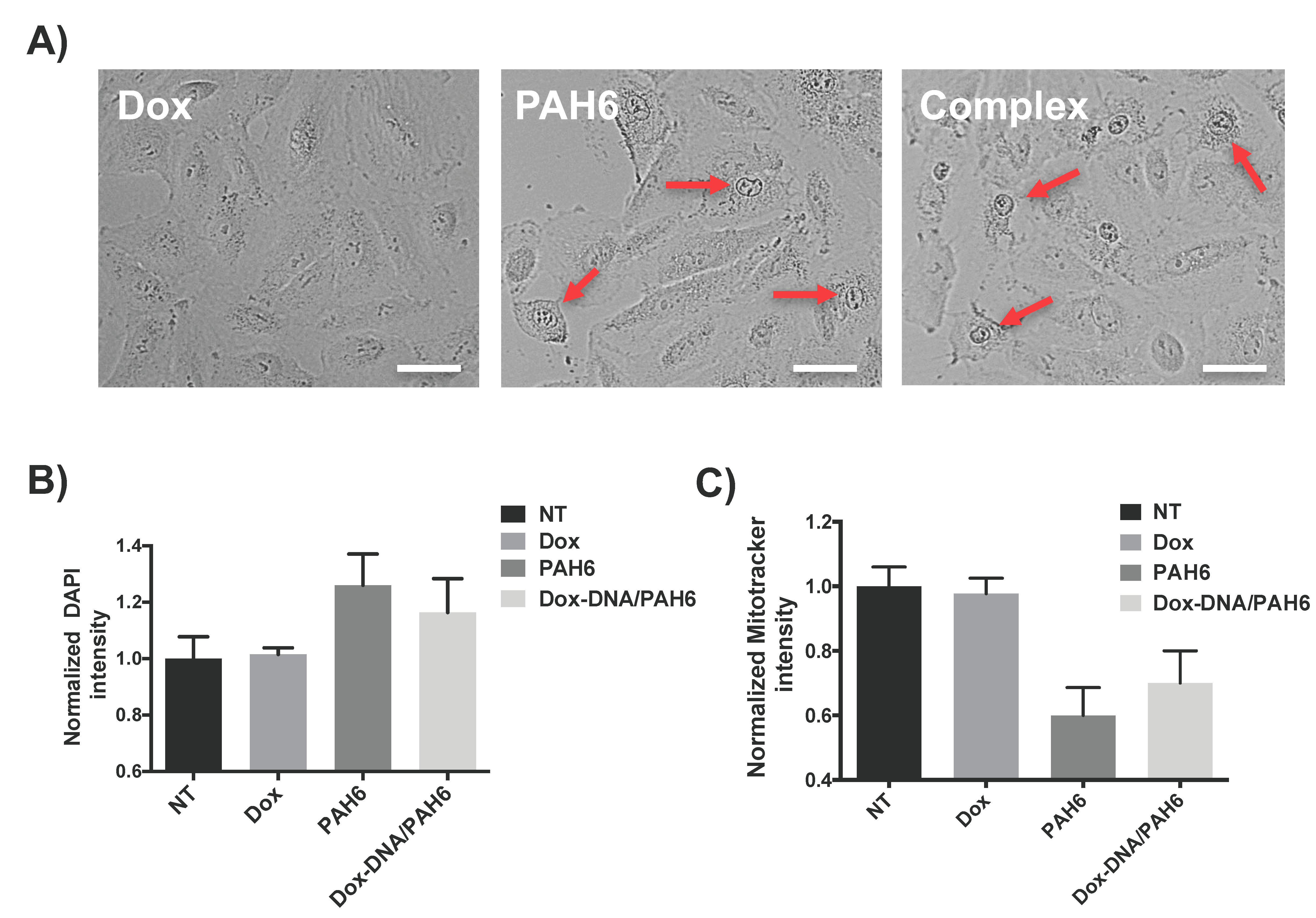
2.3. Membrane Permeabilization and Mitochondrial Depolarization Induced by PAH6
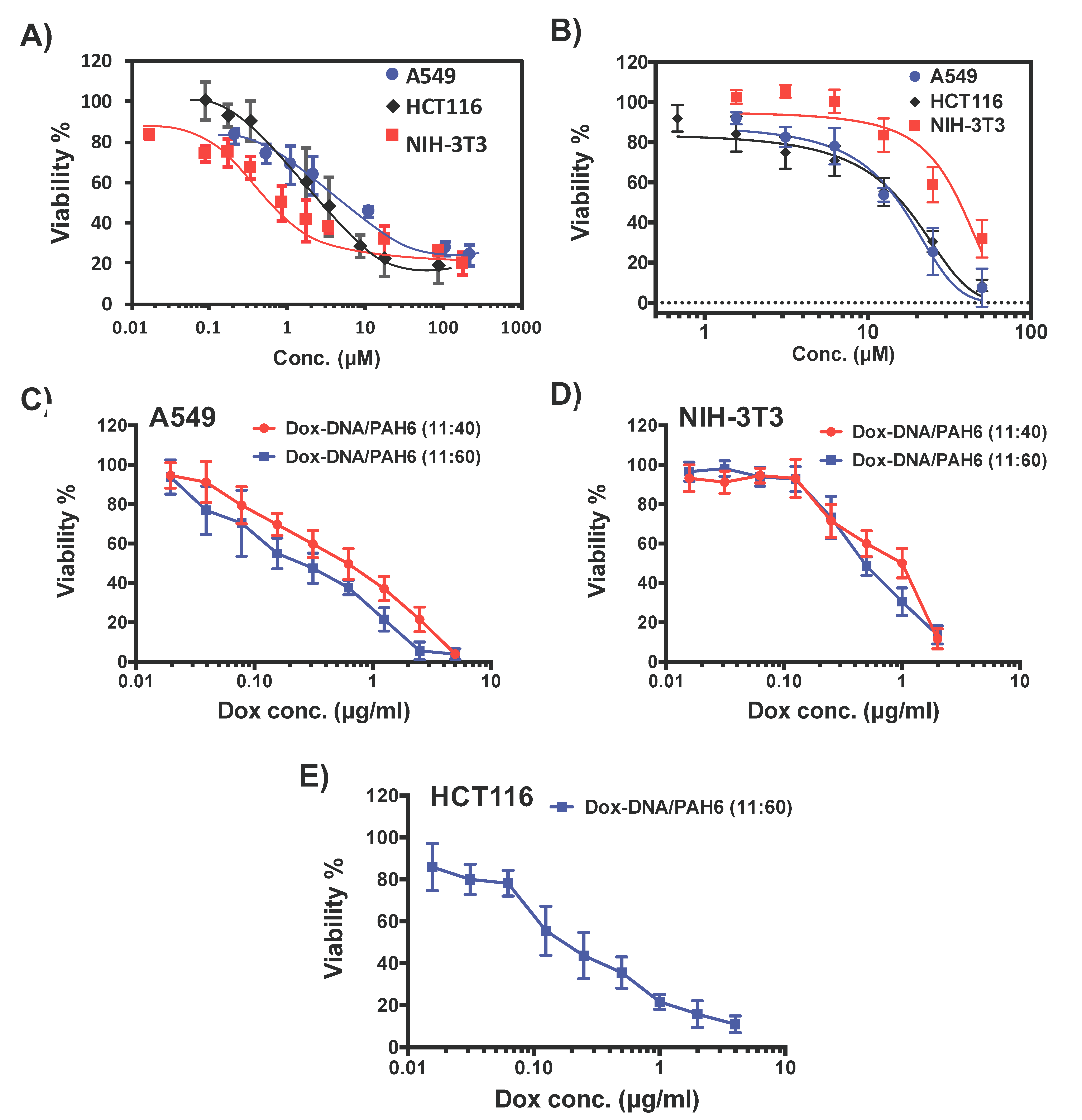
2.4. Synergistic Effect and Selectivity of the Dox-DNA/PAH6 Nanocomplex
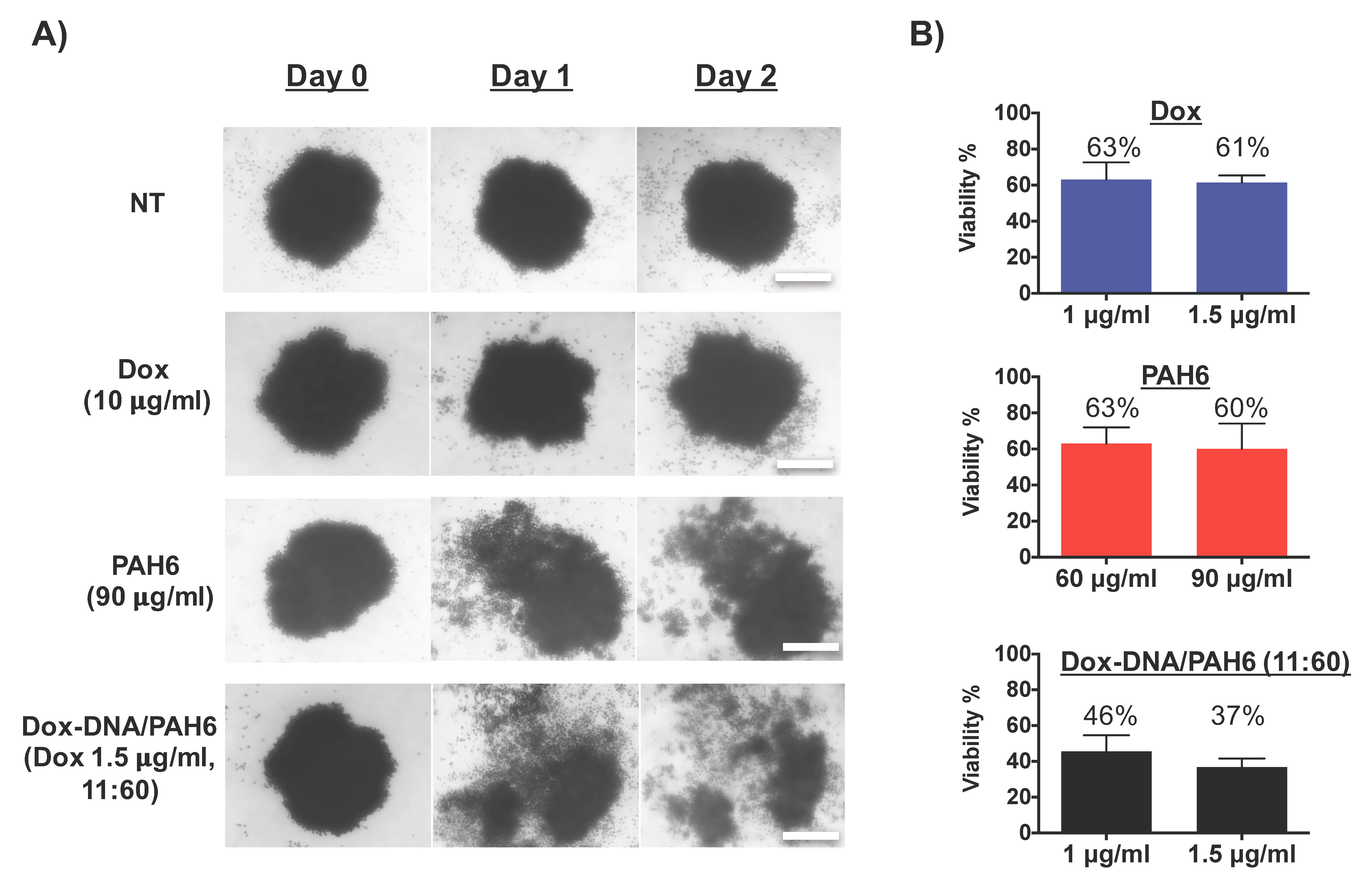
2.5. Anticancer Activity Against Lung Cancer Cell Spheroids
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation and Characterization of Dox-Loaded DNA and the Dox-DNA/PAH6 Nanocomplex
4.3. Atomic Force Microscopy (AFM)
4.4. In Vitro ATP-Triggered Dox Release
4.5. Monolayer Cell Culture
4.6. Cell Morphology Imaging
4.7. Cell Membrane Permeability Assay
4.8. Mitochondrial Membrane Potential Assay
4.9. In Vitro Cytotoxicity Test
4.10. 3D-Cultured A549 Spheroids and Anticancer Activity Test
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hu, Q.; Lin, Y.; Pacardo, D.B.; Wang, C.; Sun, W.; Ligler, F.S.; Dickey, M.D.; Gu, Z. Transformable liquid-metal nanomedicine. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Glancy, D.; Chan, W.C. DNA-controlled dynamic colloidal nanoparticle systems for mediating cellular interaction. Science 2016, 351, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The epr effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popovic, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popović, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, T.J.; Finbloom, J.A.; Chen, F.; Toft, D.J.; Cryns, V.L.; Stupp, S.I. Ph and amphiphilic structure direct supramolecular behavior in biofunctional assemblies. J. Am. Chem. Soc. 2014, 136, 14746–14752. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Bennett, W.F.; Ding, Y.; Zhang, L.; Fan, H.Y.; Zhao, D.; Zheng, T.; Ouyang, P.K.; Li, J.; Wu, Y.; et al. Design and characterization of a multifunctional ph-triggered peptide c8 for selective anticancer activity. Adv. Healthc. Mater. 2015, 4, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Jiang, T.; DiSanto, R.; Tai, W.; Gu, Z. Atp-triggered anticancer drug delivery. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, P.; Dai, Y.; Ma, P.A.; Li, X.; Cheng, Z.; Hou, Z.; Kang, X.; Li, C.; Lin, J. Multifunctional up-converting nanocomposites with smart polymer brushes gated mesopores for cell imaging and thermo/ph dual-responsive drug controlled release. Adv. Funct. Mater. 2013, 23, 4067–4078. [Google Scholar] [CrossRef]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.; Deming, T.J.; Kamei, D.T.; et al. Translocation of hiv tat peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Stanzl, E.G.; Trantow, B.M.; Vargas, J.R.; Wender, P.A. Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: Basic science, research tools, and clinical applications. Accounts Chem. Res. 2013, 46, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Perche, F.; Wang, T.; Torchilin, V.P. Matrix metalloproteinase 2-sensitive multifunctional polymeric micelles for tumor-specific co-delivery of sirna and hydrophobic drugs. Biomaterials 2014, 35, 4213–4222. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ding, Y.; Wu, Y.; Wang, R.; Pan, R.; Wan, Z.; Xu, W.; Zhang, L.; Yuan, Y.-F.; Chen, P. An amphipathic lytic peptide for enhanced and selective delivery of ellipticine. J. Mater. Chem. B 2016, 4, 4348–4355. [Google Scholar] [CrossRef]
- Allen, T.M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Tanei, T.; Godin, B.; van de Ven, A.L.; Hanibuchi, M.; Matsunoki, A.; Alexander, J.; Ferrari, M. Serum biomarkers for personalization of nanotherapeutics-based therapy in different tumor and organ microenvironments. Cancer Lett. 2014, 345, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotech. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Grantab, R.; Sivananthan, S.; Tannock, I.F. The penetration of anticancer drugs through tumor tissue as a function of cellular adhesion and packing density of tumor cells. Cancer Res. 2006, 66, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Sykes, E.A.; Chen, J.; Zheng, G.; Chan, W.C.W. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano 2014, 8, 5696–5706. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Gao, X. Functional peptides for sirna delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; El-Andaloussi, S.; Sütlü, T.; Johansson, H.; Langel, Ü. Delivery of short interfering rna using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, A.G.; Xie, H.; Coleman, D.; Glomm, W.; Ryan, J.; Anderson, M.F.; Franzen, S.; Feldheim, D.L. Multifunctional gold nanoparticle-peptide complexes for nuclear targeting. J. Am. Chem. Soc. 2003, 125, 4700–4701. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Li, Z.Y.; Zhu, J.Y.; Han, K.; Zeng, Z.Y.; Hong, W.; Li, W.X.; Jia, H.Z.; Liu, Y.; Zhuo, R.X.; et al. Dual-ph sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Standley, S.M.; Toft, D.J.; Cheng, H.; Soukasene, S.; Chen, J.; Raja, S.M.; Band, V.; Band, H.; Cryns, V.L.; Stupp, S.I. Induction of cancer cell death by self-assembling nanostructures incorporating a cytotoxic peptide. Cancer Res. 2010, 70, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Soteriou, K.; Fukumura, D.; Jain, R.K. Cationic nanoparticles have superior transvascular flux into solid tumors: Insights from a mathematical model. Ann. Biomed. Eng. 2013, 41, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Kostarelos, K.; Emfietzoglou, D.; Papakostas, A.; Yang, W.-H.; Ballangrud, Å.; Sgouros, G. Binding and interstitial penetration of liposomes within avascular tumor spheroids. Int. J. Cancer 2004, 112, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, H.; Park, S.-J.; Bae, Y.H.; Na, K. Biodegradable cationic nanoparticles loaded with an anticancer drug for deep penetration of heterogeneous tumours. Biomaterials 2013, 34, 7674–7682. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Jiang, T.; Sun, W.; Gu, Z. Atp-responsive DNA-graphene hybrid nanoaggregates for anticancer drug delivery. Biomaterials 2015, 50, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.M.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaires, J.B.; Herrera, J.; Waring, M. Preferential binding of daunomycin to 5’tacg and 5’tagc sequences revealed by footprinting titration experiments. Biochemistry 1990, 29, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Song, S.; Zhang, J.; Pan, D.; Wang, L.; Fan, C. A target-responsive electrochemical aptamer switch (treas) for reagentless detection of nanomolar atp. J. Am. Chem. Soc. 2007, 129, 1042–1043. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, T.; Han, D.; You, M.; Peng, L.; Cansiz, S.; Zhu, G.; Li, C.; Xiong, X.; Jimenez, E.; et al. Engineering of switchable aptamer micelle flares for molecular imaging in living cells. ACS Nano 2013, 7, 5724–5731. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Ji, C.; Shi, J.; Pridgen, E.M.; Frieder, J.; Wu, J.; Farokhzad, O.C. DNA self-assembly of targeted near-infrared-responsive gold nanoparticles for cancer thermo-chemotherapy. Angew. Chem. Int. Ed. 2012, 51, 11853–11857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnle, S.; Nicotera, P. Intracellular adenosine triphosphate (atp) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Shamay, Y.; Shah, J.; Isik, M.; Mizrachi, A.; Leibold, J.; Tschaharganeh, D.F.; Roxbury, D.; Budhathoki-Uprety, J.; Nawaly, K.; Sugarman, J.L.; et al. Quantitative self-assembly prediction yields targeted nanomedicines. Nat. Mater. 2018, 17, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Cui, W.; Li, J.; Sheng, YB.; Chen, P. Functional control of peptide amphiphile assemblies via modulating internal cohesion and surface chemistry switch. Chem.-Eur. J. 2018. accepted. [Google Scholar]
- Yin, N.; Ma, W.; Pei, J.; Ouyang, Q.; Tang, C.; Lai, L. Synergistic and antagonistic drug combinations depend on network topology. PLoS ONE 2014, 9, e93960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Yu, S.-c.; Ping, Y.-f.; Wu, H.; Zhao, X.; Zhang, H.; Cui, Y.; Chen, B.; Zhang, X.; Dai, J.; et al. A three-dimensional collagen scaffold cell culture system for screening anti-glioma therapeutics. Oncotarget 2016, 7, 56904–56914. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, S.; Zhao, F.; Zhang, Q.; Chen, P. Therapeutic Peptide Amphiphile as a Drug Carrier with ATP-Triggered Release for Synergistic Effect, Improved Therapeutic Index, and Penetration of 3D Cancer Cell Spheroids. Int. J. Mol. Sci. 2018, 19, 2773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092773
Lu S, Zhao F, Zhang Q, Chen P. Therapeutic Peptide Amphiphile as a Drug Carrier with ATP-Triggered Release for Synergistic Effect, Improved Therapeutic Index, and Penetration of 3D Cancer Cell Spheroids. International Journal of Molecular Sciences. 2018; 19(9):2773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092773
Chicago/Turabian StyleLu, Sheng, Feng Zhao, Qiuxin Zhang, and P. Chen. 2018. "Therapeutic Peptide Amphiphile as a Drug Carrier with ATP-Triggered Release for Synergistic Effect, Improved Therapeutic Index, and Penetration of 3D Cancer Cell Spheroids" International Journal of Molecular Sciences 19, no. 9: 2773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092773