The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
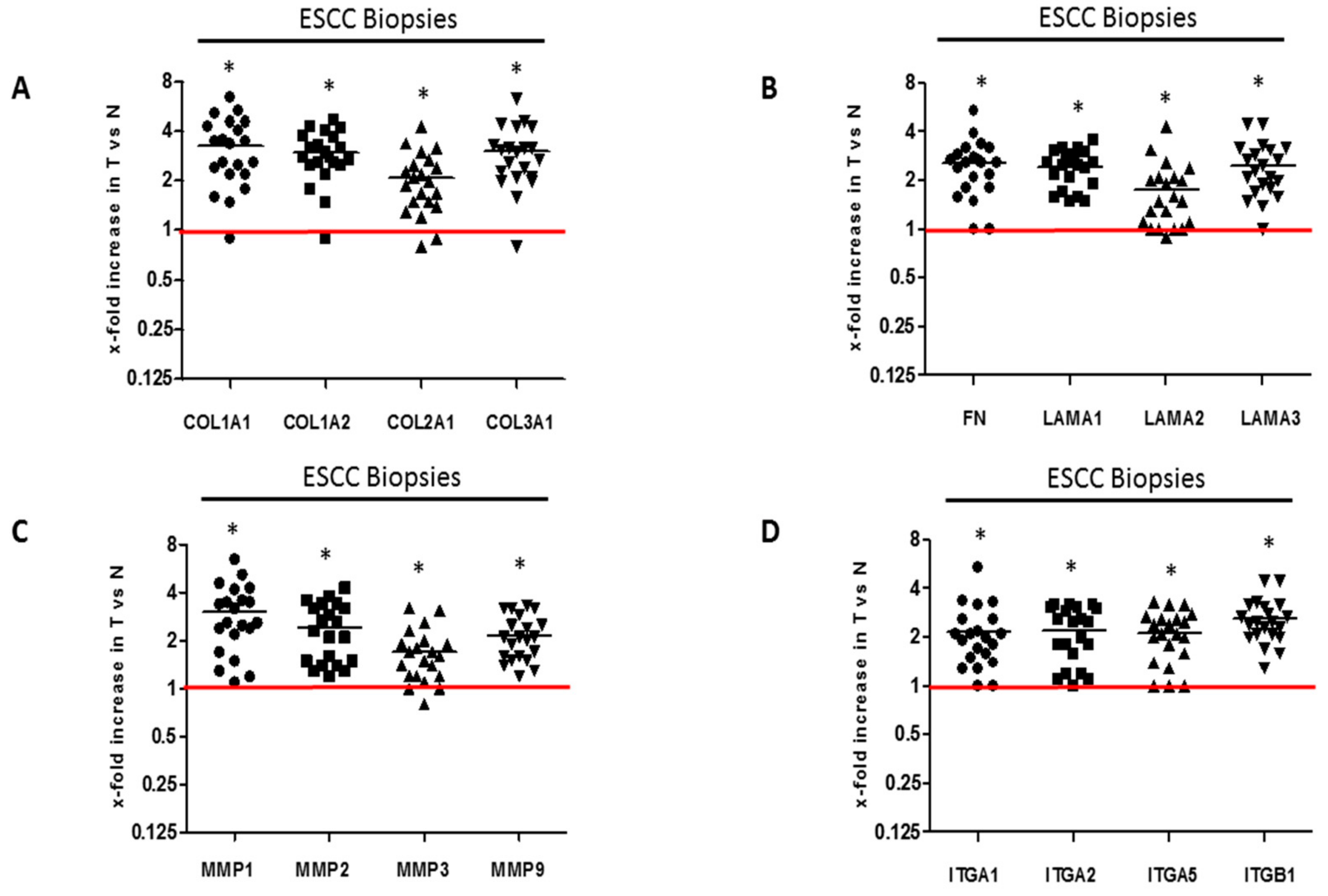
2.1. ECM Proteins and Matrix Metalloproteases Expression in Clinical Esophageal Squamous Cell Carcinoma Tissues
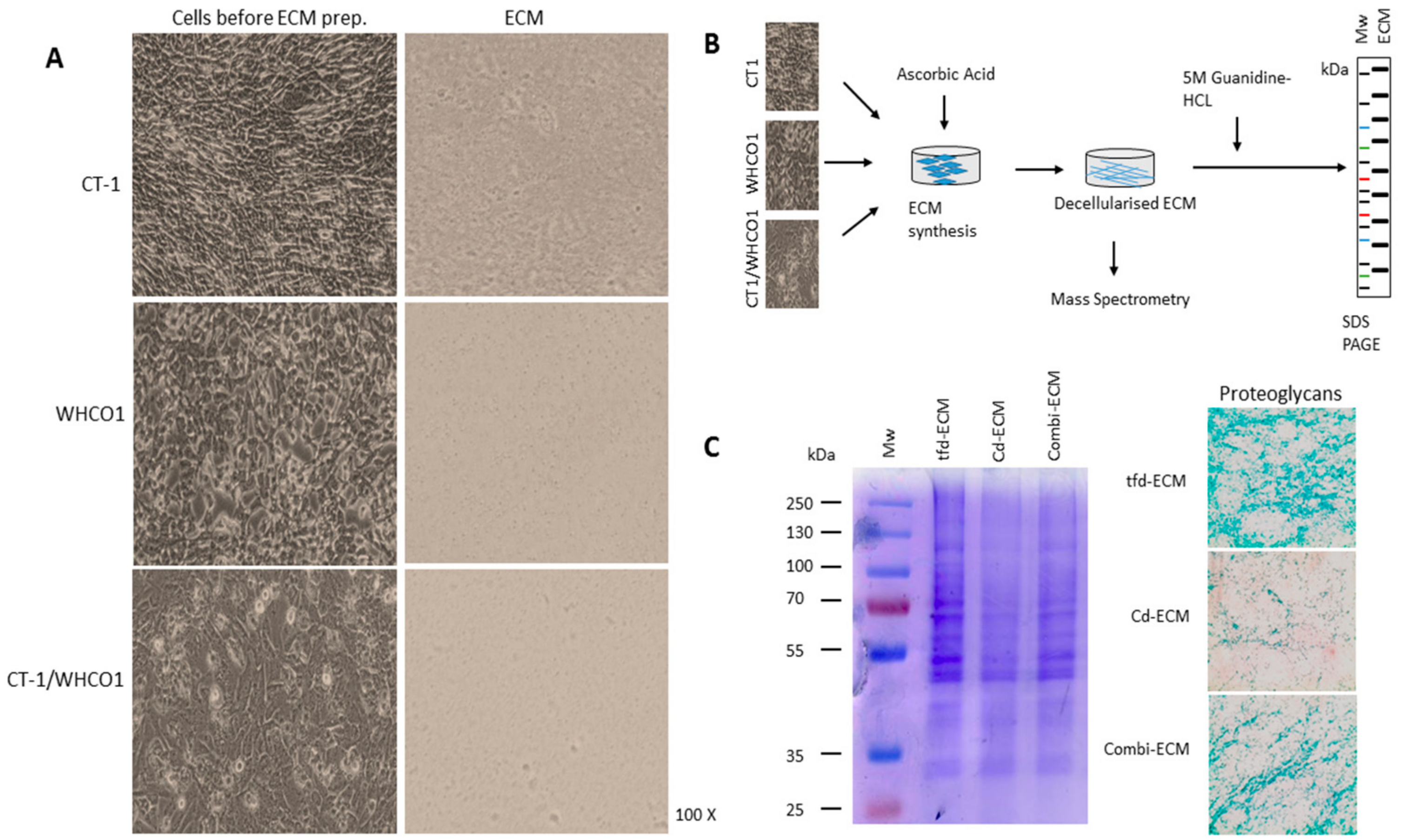
2.2. Detailed Analysis of Decellularised ECMs Used in the Study
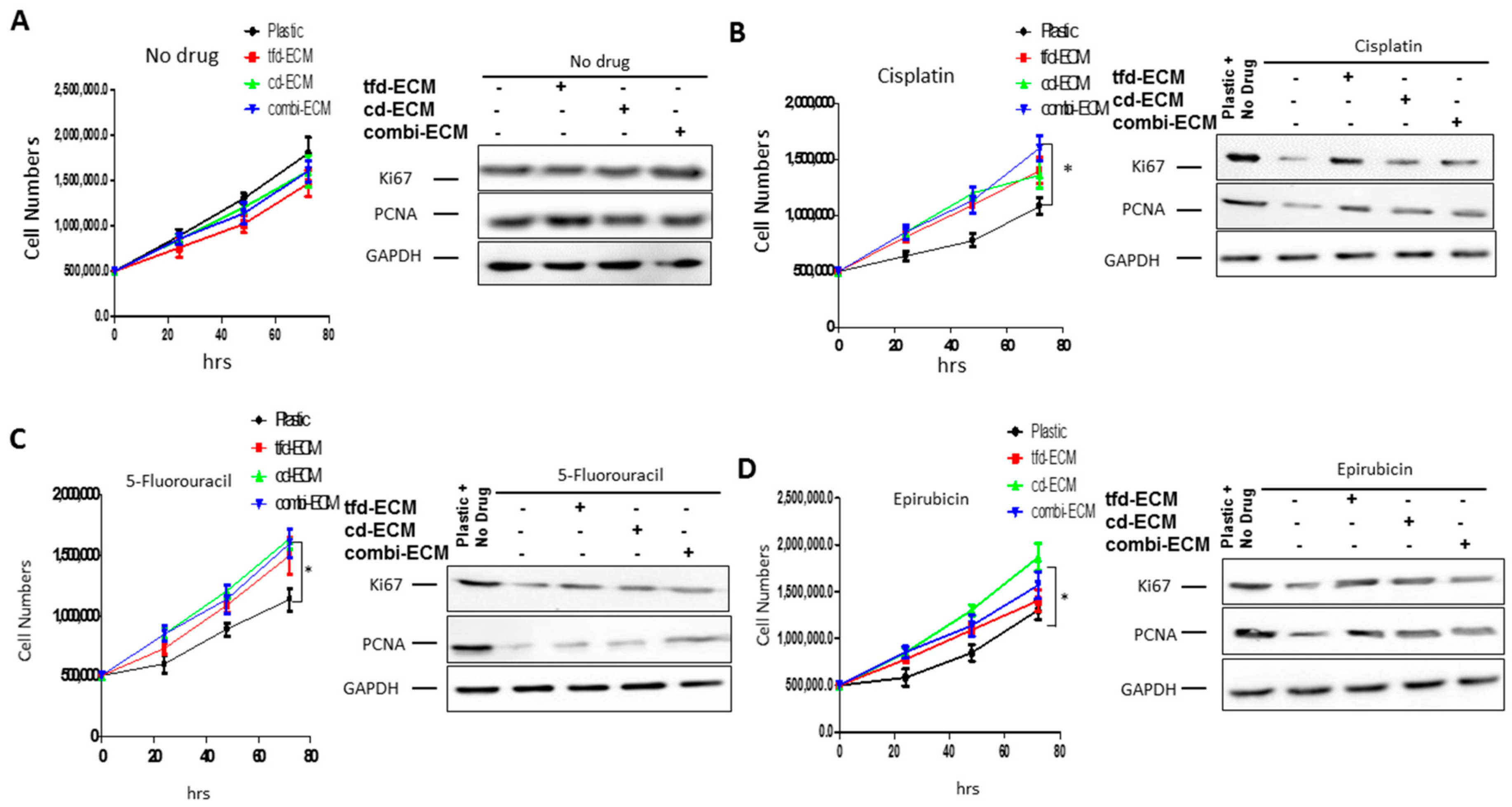
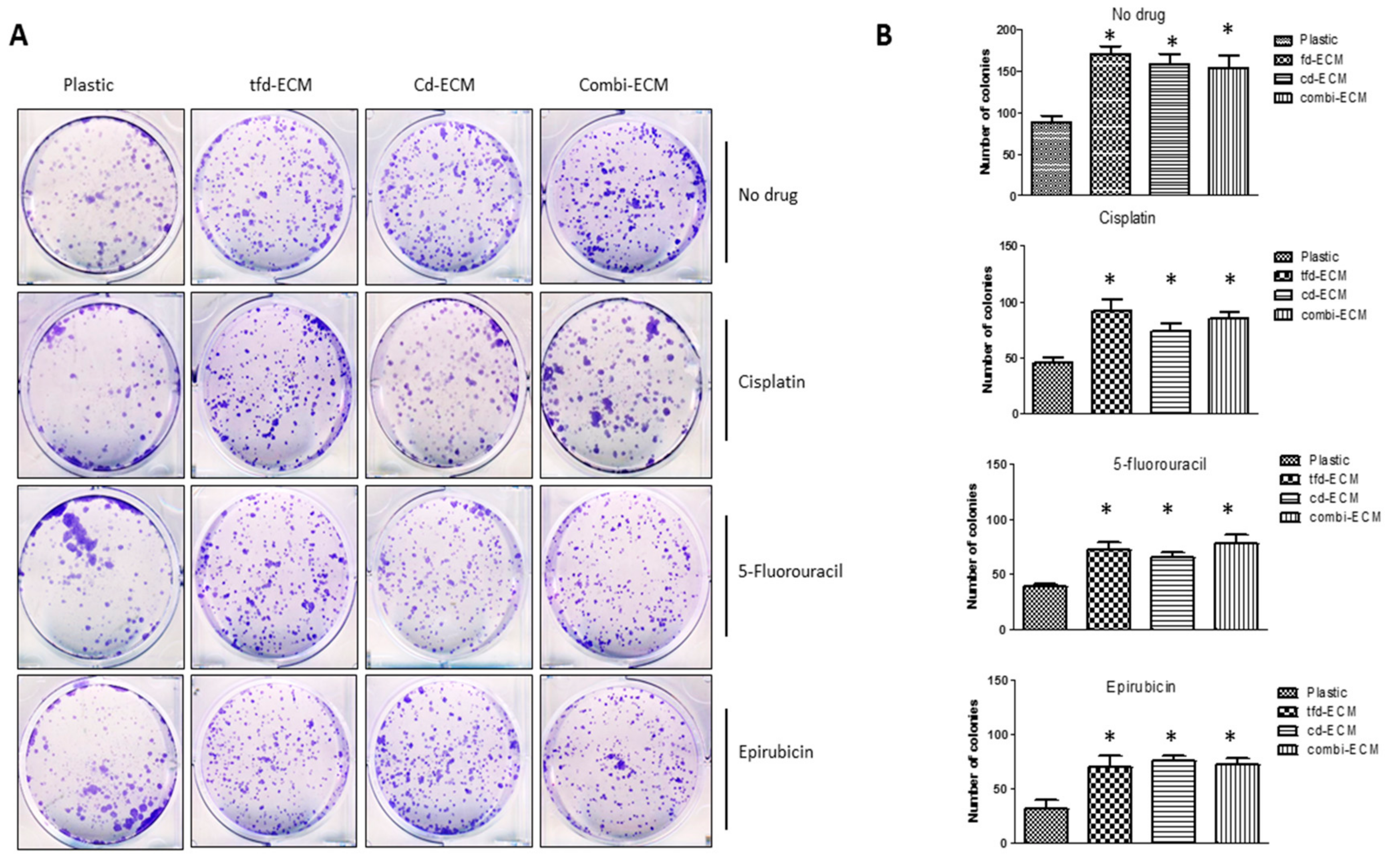
2.3. Decellularised ECMs Protect WHCO1 Cancer Cells from the Effect of Drugs
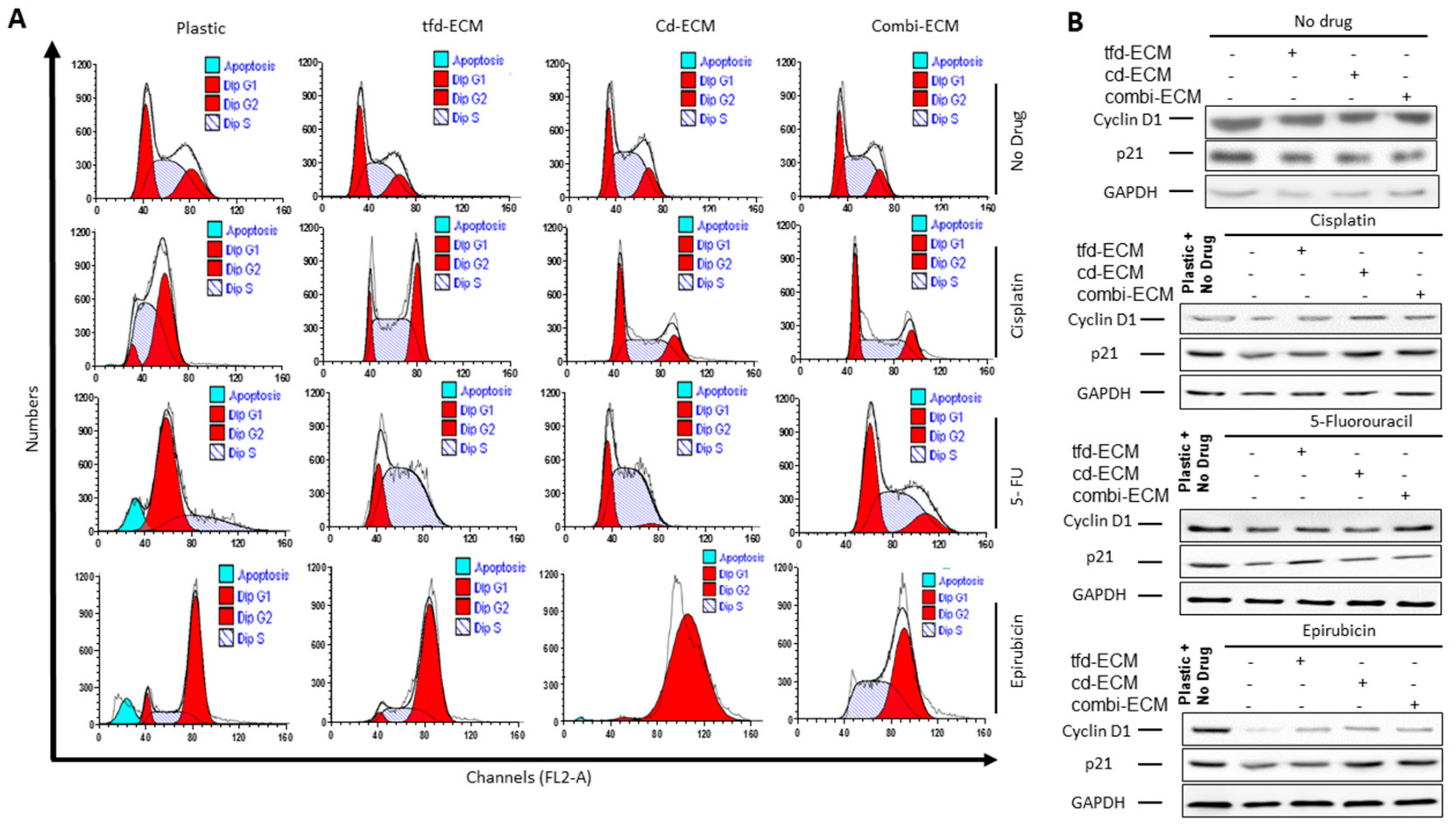
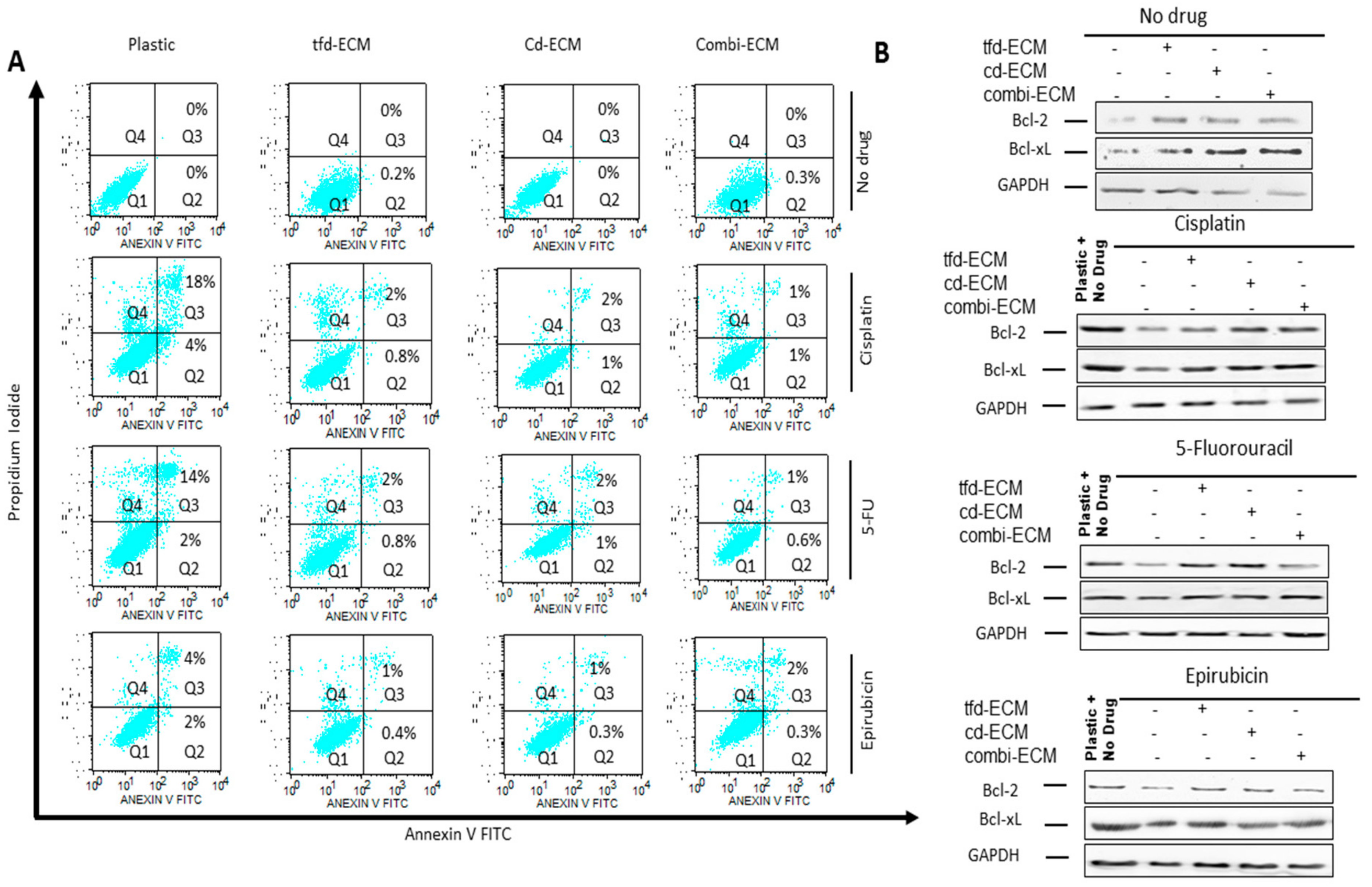
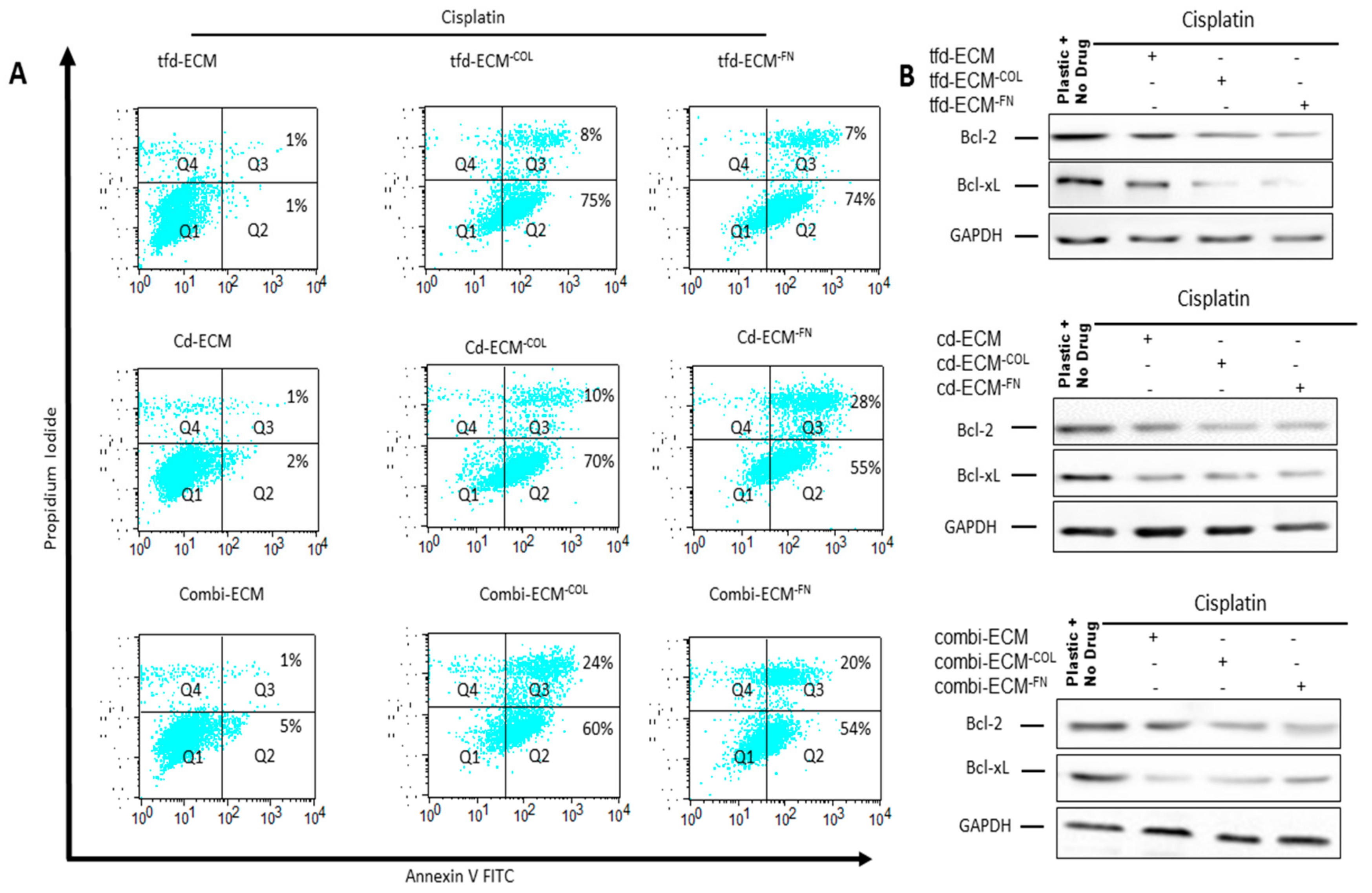
2.4. Decellularised ECMs Reduce Drug-Induced Cell Cycle Arrest and Apoptosis in WHCO1 Cancer Cells
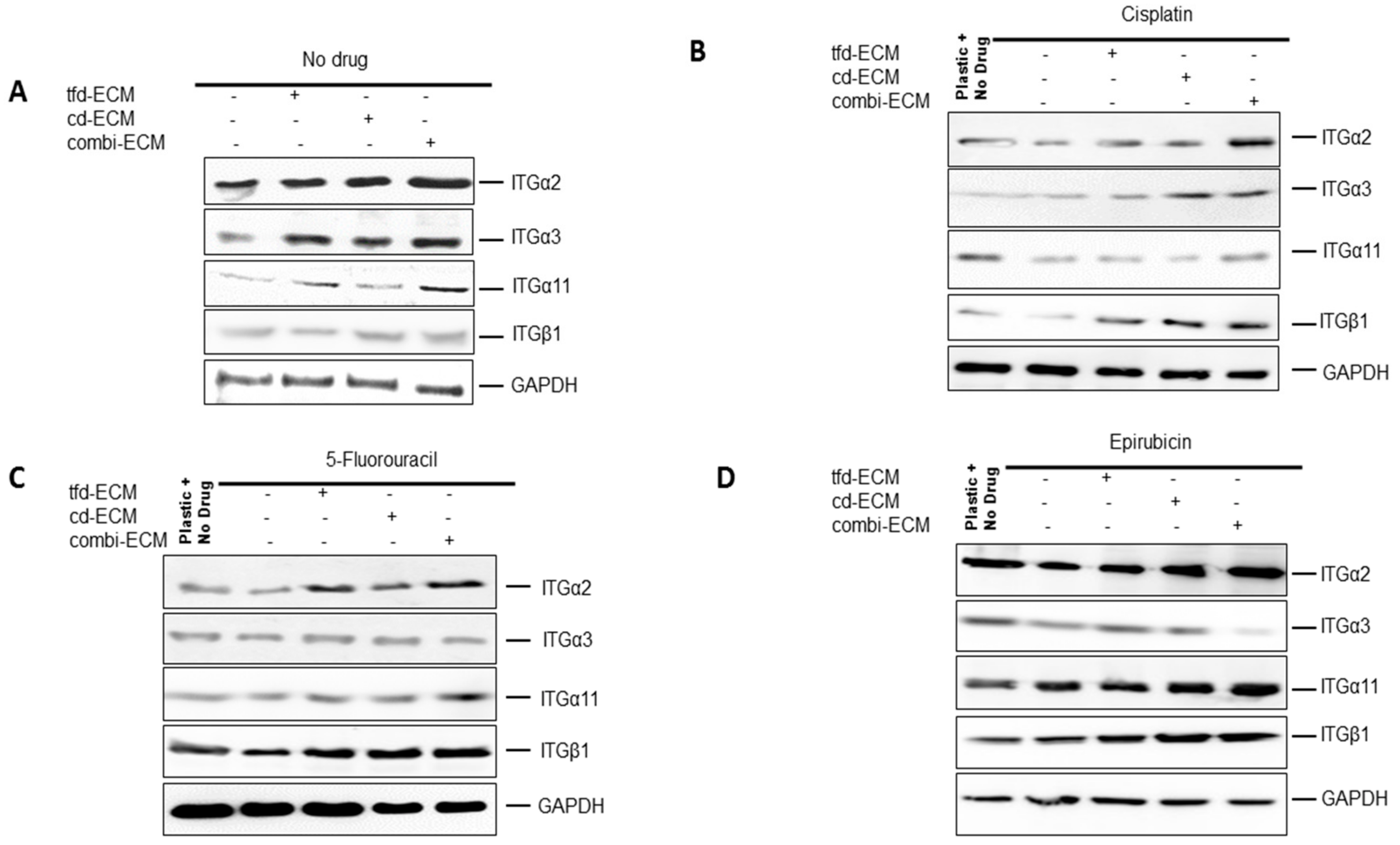
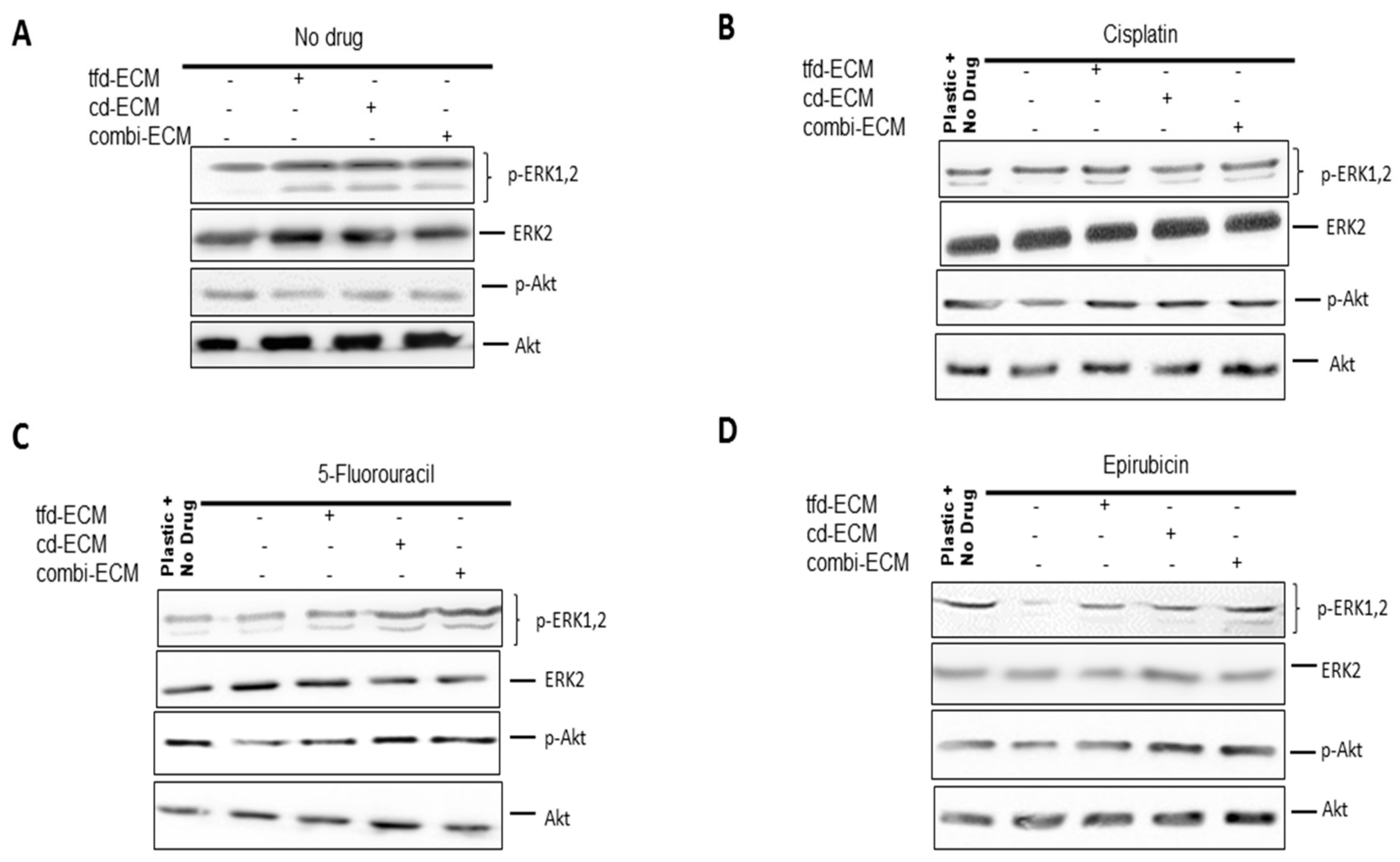
2.5. Decellularised ECMs Upregulates Several Survival Pathways in WHCO1 Cancer Cells
2.6. Type I Collagen and Fibronectin Play Key Roles in WHCO1 Cancer Cell Survival and Migration In Vitro
3. Discussion
4. Materials and Methods
4.1. Clinical Tissue Collection
4.2. Esophageal Cancer Cell Lines and Treatments
4.3. Preparation of Decellularised ECMs and ECM Coatings
4.4. Cell Cytotoxicity Assay
4.5. Quantitative Real Time RT-PCR
4.6. Immunoblot Analysis
4.7. Cell Cycle and Colony Formation Assay
4.8. Annexin V/Propidium Iodide Assay for Apoptosis
4.9. siRNA Transfection Assay
4.10. Immunofluorescence
4.11. Migration Assay
4.12. Immunohistochemistry
4.13. Mass Spectrometry
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three dimensional |
| BSA | Bovine serum albumin |
| DNA | Deoxyribonucleic acid |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetate |
| ECM | Extracellular matrix |
| ESCC | Esophageal squamous cell carcinoma |
| FN | Fibronectin |
| 5-FU | 5-fluorouracil |
| ITG | Integrin |
| Tfd-ECM | Transformed fibroblast-derived ECM |
| cd-ECM | Cancer cell-derived ECM |
| combi-ECM | Combinatorial-ECM |
| MS | Mass spectrometry. |
| MMP | Matrix metalloprotease |
| PAGE | Polyacrylamide gel electrophoresis |
| SDS | Sodium dodecyl sulphate |
References
- Bruce, A.; Evans, R.; Mezan, R.; Shi, L.; Moses, B.S.; Martin, K.H.; Gibson, L.F.; Yang, Y. Three-dimensional microfluidic tri-culture model of the bone marrow microenvironment for study of acute lymphoblastic leukemia. PLoS ONE 2015, 10, e0140506. [Google Scholar] [CrossRef] [PubMed]
- Guiro, K.; Patel, S.A.; Greco, S.J.; Rameshwar, P.; Arinzeh, T.L. Investigating breast cancer cell behavior using tissue engineering scaffolds. PLoS ONE 2015, 10, e0118724. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.H.; Park, K.M.; Ghim, J.H.; Yang, S.R.; Woo, H.M. Three dimensional culture of HEPG2 liver cells on a rat decellularized liver matrix for pharmacological studies. J. Biomed. Mater. Res. Part B Appl. Biomater. 2016, 104, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Pomo, J.M.; Taylor, R.M.; Gullapalli, R.R. Influence of TP53 and CDH1 genes in hepatocellular cancer spheroid formation and culture: A model system to understand cancer cell growth mechanics. Cancer Cell Int. 2016, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ni, C.; Grist, S.M.; Bayly, C.; Cheung, K.C. Alginate core-shell beads for simplified three-dimensional tumor spheroid culture and drug screening. Biomed. Microdevices 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L. A three-dimensional in vitro model of breast cancer: Toward replacing the need for animal experiments. Altern. Lab. Anim. ATLA 2010, 38 (Suppl. 1), 41–44. [Google Scholar] [PubMed]
- Pavlov, K.; Maley, C.C. New models of neoplastic progression in barrett’s oesophagus. Biochem. Soc. Trans. 2010, 38, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Priwitaningrum, D.L.; Blonde, J.G.; Sridhar, A.; van Baarlen, J.; Hennink, W.E.; Storm, G.; Le Gac, S.; Prakash, J. Tumor stroma-containing 3D spheroid arrays: A tool to study nanoparticle penetration. J. Control. Release 2016, 244, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Aung, K.Z.; Toh, S.L.; Goh, J.C.; Nathan, S.S. Three-dimensional porous silk tumor constructs in the approximation of in vivo osteosarcoma physiology. Biomaterials 2011, 32, 6131–6137. [Google Scholar] [CrossRef] [PubMed]
- Attieh, Y.; Vignjevic, D.M. The hallmarks of cafs in cancer invasion. Eur. J. Cell Biol. 2016, 95, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.M.; Jones, D.P.; El-Hamidi, H.; Celli, J.P. ECM composition and rheology regulate growth, motility, and response to photodynamic therapy in 3D models of pancreatic ductal adenocarcinoma. Mol. Cancer Res. MCR 2017, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Chen, S.; Yuan, W.; Fan, Q.; Tian, J.; Wang, X.; Chen, L.; Zhang, X.; Wei, W.; Liu, R.; et al. Oriented collagen fibers direct tumor cell intravasation. Proc. Natl. Acad. Sci. USA 2016, 113, 11208–11213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Santangelo, A.; Castioni, N.; Portararo, P.; Gulino, A.; Botti, L.; Parenza, M.; Cappetti, B.; Orlandi, R.; et al. Mesenchymal transition of high-grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep. 2016, 17, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, R.; Mishra, D.P. Matrix reloaded: Ccn, tenascin and sibling group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Aguado, B.A.; Caffe, J.R.; Nanavati, D.; Rao, S.S.; Bushnell, G.G.; Azarin, S.M.; Shea, L.D. Extracellular matrix mediators of metastatic cell colonization characterized using scaffold mimics of the pre-metastatic niche. Acta Biomater. 2016, 33, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshiba, T.; Tanaka, M. Decellularized matrices as in vitro models of extracellular matrix in tumor tissues at different malignant levels: Mechanism of 5-fluorouracil resistance in colorectal tumor cells. Biochim. Biophys. Acta 2016, 1863, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Mazza, G.; Rombouts, K.; Rennie Hall, A.; Urbani, L.; Vinh Luong, T.; Al-Akkad, W.; Longato, L.; Brown, D.; Maghsoudlou, P.; Dhillon, A.P.; et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci. Rep. 2015, 5, 13079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzer, M.T.; Waigel, S.; Donninger, H.; Arumugam, V.; Zacharias, W.; Clark, G.; Siskind, L.J.; Soucy, P.; Beverly, L. Fibroblast-derived extracellular matrices: An alternative cell culture system that increases metastatic cellular properties. PLoS ONE 2015, 10, e0138065. [Google Scholar] [CrossRef] [PubMed]
- Shologu, N.; Szegezdi, E.; Lowery, A.; Kerin, M.; Pandit, A.; Zeugolis, D.I. Recreating complex pathophysiologies in vitro with extracellular matrix surrogates for anticancer therapeutics screening. Drug Discov. Today 2016, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Tursky, M.L.; Nekkanti, L.P.; Jenkin, G.; Kirkland, M.A.; Pande, G. Expansion of human hematopoietic stem/progenitor cells on decellularized matrix scaffolds. Curr. Protoc. Stem Cell Biol. 2016, 36, 11–16. [Google Scholar]
- Xiong, G.; Flynn, T.J.; Chen, J.; Trinkle, C.; Xu, R. Development of an ex vivo breast cancer lung colonization model utilizing a decellularized lung matrix. Integr. Biol. Quant. Biosci. Nano Macro 2015, 7, 1518–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachman, H.; Nicosia, J.; Dysart, M.; Barker, T.H. Utilizing fibronectin integrin-binding specificity to control cellular responses. Adv. Wound Care 2015, 4, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Belair, D.G.; Le, N.N.; Murphy, W.L. Design of growth factor sequestering biomaterials. Chem. Commun. 2014, 50, 15651–15668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, D.; Creemers, E.E.; Kassiri, Z. Matrix as an interstitial transport system. Circ. Res. 2014, 114, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Hudalla, G.A.; Murphy, W.L. Chemically well-defined self-assembled monolayers for cell culture: Toward mimicking the natural ECM. Soft Matter 2011, 7, 9561–9571. [Google Scholar] [CrossRef] [PubMed]
- Hudalla, G.A.; Murphy, W.L. Biomaterials that regulate growth factor activity via bioinspired interactions. Adv. Funct. Mater. 2011, 21, 1754–1768. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Leck, K.J.; Gao, S.; Wan, A.C. Three-dimensional reconstituted extracellular matrix scaffolds for tissue engineering. Biomaterials 2009, 30, 4309–4317. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobin, A.S.; West, J.L. Effects of epidermal growth factor on fibroblast migration through biomimetic hydrogels. Biotechnol. Prog. 2003, 19, 1781–1785. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choe, C.; Shin, Y.S.; Jeon, M.J.; Choi, S.J.; Lee, J.; Bae, G.Y.; Cha, H.J.; Kim, J. Human lung cancer-associated fibroblasts enhance motility of non-small cell lung cancer cells in co-culture. Anticancer. Res. 2013, 33, 2001–2009. [Google Scholar] [PubMed]
- Morrissey, C.; Vessella, R.L. The role of tumor microenvironment in prostate cancer bone metastasis. J. Cell. Biochem. 2007, 101, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Uchinaka, A.; Tasaka, K.; Mizuno, Y.; Maeno, Y.; Ban, T.; Mori, S.; Hamada, Y.; Miyagawa, S.; Saito, A.; Sawa, Y.; et al. Laminin alpha2-secreting fibroblasts enhance the therapeutic effect of skeletal myoblast sheets. Eur. J. Cardiothorac. Surg. 2016, 51, 457–464. [Google Scholar]
- Wernert, N. The multiple roles of tumor stroma. Virchows Arch. Int. J. Pathol. 1997, 430, 433–443. [Google Scholar] [CrossRef]
- Wight, T.N.; Potter-Perigo, S. The extracellular matrix: An active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G950–G955. [Google Scholar] [CrossRef] [PubMed]
- Catteau, X.; Simon, P.; Buxant, F.; Noel, J.C. Expression of the glucocorticoid receptor in breast cancer-associated fibroblasts. Mol. Clin. Oncol. 2016, 5, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domogauer, J.D.; de Toledo, S.M.; Azzam, E.I. A mimic of the tumor microenvironment: A simple method for generating enriched cell populations and investigating intercellular communication. J. Vis. Exp. JoVE 2016. [Google Scholar] [CrossRef] [PubMed]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Serge, A.; Lavaut, M.N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, J.S.; Ligon, L.A. Stiffened extracellular matrix and signaling from stromal fibroblasts via osteoprotegerin regulate tumor cell invasion in a 3-D tumor in situ model. Cancer Microenviron. 2016, 9, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of msc with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Merlino, G.; Miodini, P.; Paolini, B.; Carcangiu, M.L.; Gennaro, M.; Dugo, M.; Daidone, M.G.; Cappelletti, V. Stromal activation by tumor cells: An in vitro study in breast cancer. Microarrays 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent carcinoma-associated fibroblasts upregulate IL8 to enhance pro-metastatic phenotypes. Mol. Cancer Res. MCR 2016, 15, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, K.; Wickett, R.R.; Kadekaro, A.L.; Zhang, Y. Targeted deactivation of cancer-associated fibroblasts by beta-catenin ablation suppresses melanoma growth. Tumour Biol. 2016, 37, 14235–14248. [Google Scholar] [CrossRef] [PubMed]
- Akrish, S.J.; Rachmiel, A.; Sabo, E.; Vered, M.; Ben-Izhak, O. Cancer associated fibroblasts are an infrequent finding in the microenvironment of proliferative verrucous leukoplakia associated squamous cell carcinoma. J. Oral Pathol. Med. 2016, 46, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.; Gillen, A.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.; Elias, A.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2016, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Epigenetic control of the tumor microenvironment. Epigenomics 2016, 8, 1671–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarty, K.; Friedman, D.; Cottam, B.; Newell, P.; Gough, M.; Crittenden, M.R.; Young, K.H. Targeting cancer-associated fibroblasts in combination with radiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, E594–E595. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2016, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, S.; Miyamoto, S.; Kikuchi, O.; Goto, T.; Amanuma, Y.; Muto, M. Recent advances from basic and clinical studies of esophageal squamous cell carcinoma. Gastroenterology 2015, 149, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.; Geller, A.; Bender, C.E.; Gostout, C.J.; Donohue, J.H. Surgical and interventional palliative treatment of upper gastrointestinal malignancies. Eur. J. Gastroenterol. Hepatol. 2000, 12, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Nash, C.L.; Gerdes, H. Methods of palliation of esophageal and gastric cancer. Surg. Oncol. Clin. N. Am. 2002, 11, 459–483. [Google Scholar] [CrossRef]
- Nojilana, B.; Bradshaw, D.; Pillay-van Wyk, V.; Msemburi, W.; Laubscher, R.; Somdyala, N.I.; Joubert, J.D.; Groenewald, P.; Dorrington, R.E. Emerging trends in non-communicable disease mortality in South Africa, 1997–2010. S. Afr. Med. J. 2016, 106, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Somdyala, N.I.; Bradshaw, D.; Gelderblom, W.C.; Parkin, D.M. Cancer incidence in a rural population of South Africa, 1998–2002. Int. J. Cancer 2010, 127, 2420–2429. [Google Scholar] [CrossRef] [PubMed]
- Best, L.M.; Mughal, M.; Gurusamy, K.S. Non-surgical versus surgical treatment for oesophageal cancer. Cochrane Database Syst. Rev. 2016, 3, CD011498. [Google Scholar] [PubMed]
- Gao, P.; Tsai, C.; Yang, Y.; Xu, Y.; Zhang, C.; Zhang, C.; Wang, L.; Liu, H.; Wang, Z. Intraoperative radiotherapy in gastric and esophageal cancer: Meta-analysis of long-term outcomes and complications. Minerva Med. 2016, 108, 74–83. [Google Scholar] [PubMed]
- Ge, L.; Wang, H.J.; Yin, D.; Lei, C.; Zhu, J.F.; Cai, X.H.; Zhang, G.Q. Effectiveness of 5-flurouracil-based neoadjuvant chemotherapy in locally-advanced gastric/gastroesophageal cancer: A meta-analysis. World J. Gastroenterol. 2012, 18, 7384–7393. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Sasaki, R.; Ejima, Y.; Ogura, M.; Negoro, Y.; Nakajima, T.; Murakami, M.; Kaji, Y.; Sugimura, K. Efficacy of intraoperative radiotherapy targeted to the abdominal lymph node area in patients with esophageal carcinoma. J. Radiat. Res. 2012, 53, 882–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.W.; Guo, Y.M.; Zhang, Q.; Fu, S. Benefits from adjuvant intraoperative radiotherapy treatment for gastric cancer: A meta-analysis. Mol. Clin. Oncol. 2015, 3, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Yang, Q.; Xie, X.F.; Yang, X.Z.; Zhang, M.Y.; Wang, H.Y.; Xu, G.L. Clinical significance and prognostic value of trim24 expression in esophageal squamous cell carcinoma. Aging 2016, 8, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Depypere, L.; Lerut, T.; Moons, J.; Coosemans, W.; Decker, G.; Van Veer, H.; De Leyn, P.; Nafteux, P. Isolated local recurrence or solitary solid organ metastasis after esophagectomy for cancer is not the end of the road. Dis. Esophagus 2017, 30, 1–8. [Google Scholar] [PubMed]
- Khan, M.; Muzaffar, A.; Syed, A.A.; Khatak, S.; Khan, A.R.; Ashraf, M.I. Changes in oncological outcomes: Comparison of the conventional and minimally invasive esophagectomy, a single institution experience. Updat. Surg. 2016, 68, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Anfossi, S.; Zheng, Y.; Cai, M.; Fu, J.; Qiu, B.; Yang, H.; Liu, Q.; Fu, J.; Liu, M.; et al. Clinical and biological prognostic factors for locoregional recurrence in patients with thoracic esophageal squamous cell carcinoma treated with radical 2-field lymph node dissection: Results from long-term follow-up. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, E175. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Z.; Zhang, G.; Zhu, Q.; Zeng, H.; Wang, T.; Gao, F.; Qi, Z.; Zhang, J.; Wang, R. High TRAF6 expression is associated with esophageal carcinoma recurrence and prompts cancer cell invasion. Oncol. Res. 2017, 25, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Hamano, R.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Hara, J.; Moon, J.H.; Nakajima, K.; Takiguchi, S.; Fujiwara, Y.; Mori, M.; et al. Overexpression of mir-200c induces chemoresistance in esophageal cancers mediated through activation of the akt signaling pathway. Clin. Cancer Res. 2011, 17, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Herbst, A.; Kalinski, T.; Qin, J.; Wang, X.; Jiang, Z.; Benedix, F.; Franke, S.; Wartman, T.; et al. Mir-221 mediates chemoresistance of esophageal adenocarcinoma by direct targeting of DKK2 expression. Ann. Surg. 2016, 264, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Xi, R.; Pan, S.; Chen, X.; Hui, B.; Zhang, L.; Fu, S.; Li, X.; Zhang, X.; Gong, T.; Guo, J.; et al. HPV16 E6-E7 induces cancer stem-like cells phenotypes in esophageal squamous cell carcinoma through the activation of Pi3K/AKT signaling pathway in vitro and in vivo. Oncotarget 2016, 7, 57050–57065. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, X.D.; Zhou, Y.; Ban, X.; Zeng, T.T.; Li, L.; Zhang, B.Z.; Yun, J.; Xie, D.; Guan, X.Y.; et al. Stemness and chemotherapeutic drug resistance induced by EIF5A2 overexpression in esophageal squamous cell carcinoma. Oncotarget 2015, 6, 26079–26089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.F.; Wu, C.; Alshareef, A.; Gupta, N.; Zhao, Q.; Xu, X.E.; Jiao, J.W.; Li, E.M.; Xu, L.Y.; Lai, R. The PI3K/AKT/C-MYC axis promotes the acquisition of cancer stem-like features in esophageal squamous cell carcinoma. Stem Cells 2016, 34, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Quaroni, L.; Casson, A.G. Identification and characterization of stemlike cells in human esophageal adenocarcinoma and normal epithelial cell lines. J. Thorac. Cardiovasc. Surg. 2012, 144, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.Y.; Yeo, S.Y.; Xuan, Y.H.; Kim, S.H. The prognostic significance of cancer-associated fibroblasts in esophageal squamous cell carcinoma. PLoS ONE 2014, 9, e99955. [Google Scholar] [CrossRef] [PubMed]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef] [PubMed]
- Jomrich, G.; Jesch, B.; Birner, P.; Schwameis, K.; Paireder, M.; Asari, R.; Schoppmann, S.F. Stromal expression of carbonic anhydrase ix in esophageal cancer. Clin. Transl. Oncol. 2014, 16, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, I.; Freudenberger, T.; Twarock, S.; Yamaguchi, Y.; Grandoch, M.; Fischer, J.W. Esophageal squamous cell carcinoma cells modulate chemokine expression and hyaluronan synthesis in fibroblasts. J. Biol. Chem. 2016, 291, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Morishima, K.; Ui, T.; Matsubara, D.; Tamura, T.; Oguni, S.; Hosoya, Y.; Sata, N.; Lefor, A.T.; Yasuda, Y.; et al. Stromal fibroblasts are predictors of disease-related mortality in esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, G.; Wang, J.; Wang, L.; Huang, X.; Cheng, Y. The role of cancer-associated fibroblasts in esophageal cancer. J. Transl. Med. 2016, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Radwanska, A.; Beghelli-de la Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-induced dormancy and chemoresistance in a549 human lung carcinoma cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumor Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, J.A.; Kuperwasser, C. Stromal biomarkers in breast cancer development and progression. Clin. Exp. Metastasis 2012, 29, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Voiles, L.; Lewis, D.E.; Han, L.; Lupov, I.P.; Lin, T.L.; Robertson, M.J.; Petrache, I.; Chang, H.C. Overexpression of type vi collagen in neoplastic lung tissues. Oncol. Rep. 2014, 32, 1897–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, G.; Deng, L.; Zhu, J.; Rychahou, P.G.; Xu, R. Prolyl-4-hydroxylase alpha subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xiong, G.; Trinkle, C.; Xu, R. Integrated extracellular matrix signaling in mammary gland development and breast cancer progression. Histol. Histopathol. 2014, 29, 1083–1092. [Google Scholar] [PubMed]
- Troester, M.A.; Lee, M.H.; Carter, M.; Fan, C.; Cowan, D.W.; Perez, E.R.; Pirone, J.R.; Perou, C.M.; Jerry, D.J.; Schneider, S.S. Activation of host wound responses in breast cancer microenvironment. Clin. Cancer Res. 2009, 15, 7020–7028. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.; Puleo, J.; Llave, A.; Mouneimne, G.; Kamm, R.D.; Nikkhah, M. Breast cancer cell invasion into a three dimensional tumor-stroma microenvironment. Sci. Rep. 2016, 6, 34094. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Turnley, T.; Wishart, A.; Rowe, A.; Kallmeyer, K.; van Vollenstee, F.A.; Thomford, N.E.; Dandara, C.; Chopera, D.; Pepper, M.S.; et al. Fibroblast-derived extracellular matrix induces chondrogenic differentiation in human adipose-derived mesenchymal stromal/stem cells in vitro. Int. J. Mol. Sci. 2016, 17, 1259. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Vogelsang, M.; Thomford, N.E.; Dandara, C.; Kallmeyer, K.; Pepper, M.S.; Parker, M.I. Wharton’s jelly-derived mesenchymal stromal cells and fibroblast-derived extracellular matrix synergistically activate apoptosis in a p21-dependent mechanism in WHCO1 and MDA MB 231 cancer cells in vitro. Stem Cells Int. 2016, 2016, 4842134. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.; Conway, J.R.; Vennin, C.; Magenau, A.; Hughes, W.E.; Morton, J.P.; Timpson, P. Three-dimensional cancer models mimic cell-matrix interactions in the tumor microenvironment. Carcinogenesis 2014, 35, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Serebriiskii, I.; Castello-Cros, R.; Lamb, A.; Golemis, E.A.; Cukierman, E. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2008, 27, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namba, M.; Nishitani, K.; Kimoto, T. Effects of theophylline on the cell growth of normal and malignant human cells transformed in culture. Gan 1980, 71, 621–627. [Google Scholar] [PubMed]
- Bao, C.H.; Wang, X.T.; Ma, W.; Wang, N.N.; Un Nesa, E.; Wang, J.B.; Wang, C.; Jia, Y.B.; Wang, K.; Tian, H.; et al. Irradiated fibroblasts promote epithelial-mesenchymal transition and hdgf expression of esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2015, 458, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, P.L.; Russell, P.; Scott, A.M.; John, T. Targeting the vasculature: Anti-angiogenic agents for malignant mesothelioma. Expert Rev. Anticancer. Ther. 2016, 16, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Chiron, D.; Bellanger, C.; Papin, A.; Tessoulin, B.; Dousset, C.; Maiga, S.; Moreau, A.; Esbelin, J.; Trichet, V.; Chen-Kiang, S.; et al. Microenvironment-dependent proliferation and mitochondrial priming loss in mantle cell lymphoma is overcome by anti-CD20. Blood 2016, 128, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Samols, M.A.; Morsberger, L.A.; Yonescu, R.; Thiess, M.L.; Batista, D.A.; Ning, Y.; Burns, K.H.; Vuica-Ross, M.; Borowitz, M.J.; et al. Tumor-infiltrating macrophages in post-transplant, relapsed classical hodgkin lymphoma are donor-derived. PLoS ONE 2016, 11, e0163559. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Brown, H.; Holen, I. The endocrine influence on the bone microenvironment in early breast cancer. Endocr. Relat. Cancer 2016, 23, R567–R576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, C.; Wang, M.; Hou, H.; Cui, L.; Jiang, C.; Sun, J.; Qu, X. Prognostic significance of immune cells in the tumor microenvironment and peripheral blood of gallbladder carcinoma patients. Clin. Transl. Oncol. 2016, 19, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y. What are the hallmarks of cancer? Nat. Rev. Cancer 2010, 10, 232–233. [Google Scholar] [CrossRef] [PubMed]
- Tommelein, J.; Verset, L.; Boterberg, T.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front. Oncol. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wintzell, M.; Hjerpe, E.; Avall Lundqvist, E.; Shoshan, M. Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 2012, 12, 359. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [PubMed]
- Bourguignon, L.Y. Matrix hyaluronan promotes specific microrna upregulation leading to drug resistance and tumor progression. Int. J. Mol. Sci. 2016, 17, 517. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Shiina, M.; Li, J.J. Hyaluronan-cd44 interaction promotes oncogenic signaling, microrna functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv. Cancer Res. 2014, 123, 255–275. [Google Scholar] [PubMed]
- Bulysheva, A.A.; Bowlin, G.L.; Petrova, S.P.; Yeudall, W.A. Enhanced chemoresistance of squamous carcinoma cells grown in 3D cryogenic electrospun scaffolds. Biomed. Mater. 2013, 8, 055009. [Google Scholar] [CrossRef] [PubMed]
- Le Calve, B.; Griveau, A.; Vindrieux, D.; Marechal, R.; Wiel, C.; Svrcek, M.; Gout, J.; Azzi, L.; Payen, L.; Cros, J.; et al. Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution. Oncotarget 2016, 7, 32100–32112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, L.S.; Huang, P.H. The pathobiology of collagens in glioma. Mol. Cancer Res. MCR 2013, 11, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Biswas, A.; Mandal, M. Glioma progression through the prism of heat shock protein mediated extracellular matrix remodeling and epithelial to mesenchymal transition. Exp. Cell Res. 2017, 359, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Kohi, S.; Hirata, K.; Goggins, M. Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. 2016, 107, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Clementz, A.G.; Harris, A. Collagen xv: Exploring its structure and role within the tumor microenvironment. Mol. Cancer Res. MCR 2013, 11, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. Lox-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.; Zhang, M.; Qiu, Q.; Skuli, N.; Nakazawa, M.S.; Karakasheva, T.; Mucaj, V.; Shay, J.E.; Stangenberg, L.; Sadri, N.; et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013, 3, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013, 73, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Goto, R.; Nakamura, Y.; Takami, T.; Sanke, T.; Tozuka, Z. Quantitative lc-ms/ms analysis of proteins involved in metastasis of breast cancer. PLoS ONE 2015, 10, e0130760. [Google Scholar] [CrossRef] [PubMed]
- Raglow, Z.; Thomas, S.M. Tumor matrix protein collagen xialpha1 in cancer. Cancer Lett. 2015, 357, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, R.; Ge, L.; Zhu, Q.; Li, F. Fibrillar type I collagen matrices enhance metastasis/invasion of ovarian epithelial cancer via beta1 integrin and pten signals. Int. J. Gynecol. Cancer 2012, 22, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Spivey, K.A.; Chung, I.; Banyard, J.; Adini, I.; Feldman, H.A.; Zetter, B.R. A role for collagen xxiii in cancer cell adhesion, anchorage-independence and metastasis. Oncogene 2012, 31, 2362–2372. [Google Scholar] [CrossRef] [PubMed]
- Tanis, T.; Cincin, Z.B.; Gokcen-Rohlig, B.; Bireller, E.S.; Ulusan, M.; Tanyel, C.R.; Cakmakoglu, B. The role of components of the extracellular matrix and inflammation on oral squamous cell carcinoma metastasis. Arch. Oral Biol. 2014, 59, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Torzilli, P.A.; Bourne, J.W.; Cigler, T.; Vincent, C.T. A new paradigm for mechanobiological mechanisms in tumor metastasis. Semin. Cancer Biol. 2012, 22, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, J.; Arozarena, I.; Ehrhardt, M.; Wellbrock, C. Combination of mek and src inhibition suppresses melanoma cell growth and invasion. Oncogene 2013, 32, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Hayashido, Y.; Kitano, H.; Sakaue, T.; Fujii, T.; Suematsu, M.; Sakurai, S.; Okamoto, T. Overexpression of integrin alphav facilitates proliferation and invasion of oral squamous cell carcinoma cells via MEK/ERK signaling pathway that is activated by interaction of integrin alphavbeta8 with type collagen. Int. J. Oncol. 2014, 45, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.H.; Croteau, W.; Mullins, D.W.; Brinckerhoff, C.E. The BRAF(V600E) inhibitor, PLX4032, increases type I collagen synthesis in melanoma cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 48, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Takino, T.; Li, Z.; Domoto, T.; Sato, H. Vinculin negatively regulates transcription of mt1-mmp through MEK/ERK pathway. Biochem. Biophys. Res. Commun. 2014, 455, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Alemany-Ribes, M.; Semino, C.E. Bioengineering 3D environments for cancer models. Adv. Drug Deliv. Rev. 2014, 79–80, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Benien, P.; Swami, A. 3D tumor models: History, advances and future perspectives. Future Oncol. 2014, 10, 1311–1327. [Google Scholar] [CrossRef] [PubMed]
- Hirt, C.; Papadimitropoulos, A.; Mele, V.; Muraro, M.G.; Mengus, C.; Iezzi, G.; Terracciano, L.; Martin, I.; Spagnoli, G.C. “In vitro” 3D models of tumor-immune system interaction. Adv. Drug Deliv. Rev. 2014, 79–80, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, K.E.; Beebe, D.J. Microfluidic 3D models of cancer. Adv. Drug Deliv. Rev. 2014, 79–80, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Tanner, K.; Gottesman, M.M. Beyond 3D culture models of cancer. Sci. Transl. Med. 2015, 7, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aihara, A.; Abe, N.; Saruhashi, K.; Kanaki, T.; Nishino, T. A novel 3-D cell culture system for in vitro evaluation of anticancer drugs under anchorage-independent conditions. Cancer Sci. 2016, 107, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, K.A.; Guo, J.; Tierney, E.G.; Curtin, C.M.; Malhotra, M.; Darcy, R.; O’Brien, F.J.; O’Driscoll, C.M. The use of collagen-based scaffolds to simulate prostate cancer bone metastases with potential for evaluating delivery of nanoparticulate gene therapeutics. Biomaterials 2015, 66, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Fraley, S.I.; Wu, P.H.; He, L.; Feng, Y.; Krisnamurthy, R.; Longmore, G.D.; Wirtz, D. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci. Rep. 2015, 5, 14580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Mandal, M.; Hutmacher, D.W.; Russell, P.J.; Soekmadji, C.; Kundu, S.C. Engineered silk fibroin protein 3D matrices for in vitro tumor model. Biomaterials 2011, 32, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Stephens, C.; Walpole, C.; Swedberg, J.E.; Boyle, G.M.; Parsons, P.G.; McGuckin, M.A.; Harris, J.M.; Clements, J.A. Paclitaxel resistance and multicellular spheroid formation are induced by kallikrein-related peptidase 4 in serous ovarian cancer cells in an ascites mimicking microenvironment. PLoS ONE 2013, 8, e57056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loessner, D.; Rizzi, S.C.; Stok, K.S.; Fuehrmann, T.; Hollier, B.; Magdolen, V.; Hutmacher, D.W.; Clements, J.A. A bioengineered 3D ovarian cancer model for the assessment of peptidase-mediated enhancement of spheroid growth and intraperitoneal spread. Biomaterials 2013, 34, 7389–7400. [Google Scholar] [CrossRef] [PubMed]
- Loessner, D.; Stok, K.S.; Lutolf, M.P.; Hutmacher, D.W.; Clements, J.A.; Rizzi, S.C. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials 2010, 31, 8494–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soritau, O.; Tomuleasa, C.I.; Pall, E.; Virag, P.; Fischer-Fodor, E.; Foris, V.; Barbos, O.; Tatomir, C.; Kacso, G.; Irimie, A. Enhanced chemoresistance and tumor sphere formation as a laboratory model for peritoneal micrometastasis in epithelial ovarian cancer. Rom. J. Morphol. Embryol. 2010, 51, 259–264. [Google Scholar] [PubMed]
- Hirt, C.; Papadimitropoulos, A.; Muraro, M.G.; Mele, V.; Panopoulos, E.; Cremonesi, E.; Ivanek, R.; Schultz-Thater, E.; Droeser, R.A.; Mengus, C.; et al. Bioreactor-engineered cancer tissue-like structures mimic phenotypes, gene expression profiles and drug resistance patterns observed “in vivo”. Biomaterials 2015, 62, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Yu, S.C.; Ping, Y.F.; Wu, H.; Zhao, X.; Zhang, H.; Cui, Y.; Chen, B.; Zhang, X.; Dai, J.; et al. A three-dimensional collagen scaffold cell culture system for screening anti-glioma therapeutics. Oncotarget 2016, 7, 56904–56914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, J.; Kumar, A. Thermo-responsive polymer aided spheroid culture in cryogel based platform for high throughput drug screening. Analyst 2016, 141, 2553–2567. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Zhu, Y.X.; Ma, H.C.; Chen, S.N.; Chao, J.Y.; Ruan, W.D.; Wang, D.; Du, F.G.; Meng, Y.Z. Developing multi-cellular tumor spheroid model (MCTS) in the chitosan/collagen/alginate (CCA) fibrous scaffold for anticancer drug screening. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 62, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, J.; Zhu, L.; Lowrey, J.J.; Zhang, Y.; Hou, W.; Dong, J.; Du, Y. A ready-to-use, versatile, multiplex-able three-dimensional scaffold-based immunoassay chip for high throughput hepatotoxicity evaluation. Lab Chip 2015, 15, 2634–2646. [Google Scholar] [CrossRef] [PubMed]
- Reticker-Flynn, N.E.; Malta, D.F.; Winslow, M.M.; Lamar, J.M.; Xu, M.J.; Underhill, G.H.; Hynes, R.O.; Jacks, T.E.; Bhatia, S.N. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat. Commun. 2012, 3, 1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshiba, T.; Tanaka, M. Breast cancer cell behaviors on staged tumorigenesis-mimicking matrices derived from tumor cells at various malignant stages. Biochem. Biophys. Res. Commun. 2013, 439, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Castello-Cros, R.; Khan, D.R.; Simons, J.; Valianou, M.; Cukierman, E. Staged stromal extracellular 3D matrices differentially regulate breast cancer cell responses through PI3K and beta1-integrins. BMC Cancer 2009, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M. Q&A: Mina bissell on tumors as organs. Cancer Discov. 2013, 3, 7. [Google Scholar] [PubMed]
- Bissell, M.J. Thinking in three dimensions: Discovering reciprocal signaling between the extracellular matrix and nucleus and the wisdom of microenvironment and tissue architecture. Mol. Biol. Cell 2016, 27, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J. Goodbye flat biology-time for the 3rd and the 4th dimensions. J. Cell Sci. 2017, 130, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.X.; Qiu, J.; et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.A.; Bissell, M.J.; Lelievre, S.A. Three-dimensional culture of human breast epithelial cells: The how and the why. Methods Mol. Biol. 2013, 945, 193–219. [Google Scholar] [PubMed]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69–70, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, R.C.; Sethi, T. The role of extracellular matrix in small-cell lung cancer. Lancet. Oncol. 2001, 2, 437–442. [Google Scholar] [CrossRef]
- Rintoul, R.C.; Sethi, T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin. Sci. 2002, 102, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Ginsberg, M.H.; Downward, J.; Hughes, P.E. The small GTP-binding protein R-RAS can influence integrin activation by antagonizing a RAS/RAF-initiated integrin suppression pathway. Mol. Biol. Cell 1999, 10, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Woll, P.J. Growth factors and lung cancer. Cancer Treat. Res. 1995, 72, 111–130. [Google Scholar] [PubMed]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin alpha5beta1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Janouskova, H.; Ray, A.M.; Noulet, F.; Lelong-Rebel, I.; Choulier, L.; Schaffner, F.; Lehmann, M.; Martin, S.; Teisinger, J.; Dontenwill, M. Activation of p53 pathway by nutlin-3a inhibits the expression of the therapeutic target alpha5 integrin in colon cancer cells. Cancer Lett. 2013, 336, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Janouskova, H.; Dontenwill, M. Integrins and p53 pathways in glioblastoma resistance to temozolomide. Front. Oncol. 2012, 2, 157. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Sayeed, A.; Breen, M.; Zarif, M.J.; Garlick, D.S.; Leav, I.; Davis, R.J.; Fitzgerald, T.J.; Morrione, A.; Hsieh, C.C.; et al. Beta1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-jun amino terminal kinase 1. J. Cell. Physiol. 2013, 228, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.S.; Huang, W.H.; Liou, H.C.; Chuang, W.J.; Yang, R.S.; Fu, W.M. Upregulation of drug transporter expression by osteopontin in prostate cancer cells. Mol. Pharmacol. 2013, 83, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant PRE-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Gang, E.J.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin alpha4, TBC3486, on pre-b-all cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Dey, N.; Terakedis, B.; Bergsagel, P.L.; Li, Z.H.; Mahadevan, D.; Garlich, J.R.; Trudel, S.; Makale, M.T.; Durden, D.L. An integrin-targeted, pan-isoform, phosphoinositide-3 kinase inhibitor, SF1126, has activity against multiple myeloma in vivo. Cancer Chemother. Pharmacol. 2013, 71, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; Puig, N.; Maiso, P.; Rodriguez, I.; Alignani, D.; Burgos, L.; Sanchez, M.L.; Barcena, P.; et al. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: A new model to understand chemoresistance. Blood 2016, 127, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Carlson, J.H.; Jepperson, T.; Willis, S.; Leyland-Jones, B.; Dey, N. Rac1 GTP-ASE signals wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget 2017, 8, 3072–3103. [Google Scholar] [PubMed]
- Dhawan, A.; Friedrichs, J.; Bonin, M.V.; Bejestani, E.P.; Werner, C.; Wobus, M.; Chavakis, T.; Bornhauser, M. Breast cancer cells compete with hematopoietic stem and progenitor cells for intercellular adhesion molecule 1-mediated binding to the bone marrow microenvironment. Carcinogenesis 2016, 37, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Lee, S.O.; Doddapaneni, R.; Singh, M.; Safe, S. NR4A1 antagonists inhibit beta1-integrin-dependent breast cancer cell migration. Mol. Cell. Biol. 2016, 36, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, C.; Zhao, C.; Zhai, L.; Lv, S. Downregulation of beta3 integrin by mir-30a-5p modulates cell adhesion and invasion by interrupting erk/ets1 network in triple-negative breast cancer. Int. J. Oncol. 2016, 48, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Morozevich, G.E.; Kozlova, N.I.; Susova, O.Y.; Karalkin, P.A.; Berman, A.E. Implication of alpha2beta1 integrin in anoikis of mcf-7 human breast carcinoma cells. Biochem. Biokhimiia 2015, 80, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sarper, M.; Allen, M.D.; Gomm, J.; Haywood, L.; Decock, J.; Thirkettle, S.; Ustaoglu, A.; Sarker, S.J.; Marshall, J.; Edwards, D.R.; et al. Loss of mmp-8 in ductal carcinoma in situ (DCIS)-associated myoepithelial cells contributes to tumor promotion through altered adhesive and proteolytic function. Breast Cancer Res. BCR 2017, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Lu, Z.; Zhang, Y.; Wang, M.; Li, W.; Hu, Z.; Wang, S.; Lin, Y. Interleukin-8 upregulates integrin beta3 expression and promotes estrogen receptor-negative breast cancer cell invasion by activating the PI3K/AKT/NF-Kappab pathway. Cancer Lett. 2015, 364, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Veine, D.M.; Livant, D.L. Therapeutic inhibition of breast cancer bone metastasis progression and lung colonization: Breaking the vicious cycle by targeting alpha5beta1 integrin. Breast Cancer Res. Treat. 2016, 157, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Dickreuter, E.; Eke, I.; Krause, M.; Borgmann, K.; van Vugt, M.A.; Cordes, N. Targeting of beta1 integrins impairs DNA repair for radiosensitization of head and neck cancer cells. Oncogene 2016, 35, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.; Moran-Jones, K.; Sansom, O.J.; Brunton, V.G.; Frame, M.C. Fak deletion promotes p53-mediated induction of p21, DNA-damage responses and radio-resistance in advanced squamous cancer cells. PLoS ONE 2011, 6, e27806. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; McIntosh, J.L.; Fang, L.; Szabo, C.; Hoyt, D.G. Integrin-mediated suppression of endotoxin-induced DNA damage in lung endothelial cells is sensitive to poly(ADP-RIBOSE) polymerase-1 gene deletion. Int. J. Mol. Med. 2003, 12, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amornsupak, K.; Insawang, T.; Thuwajit, P.; O-Charoenrat, P.; Eccles, S.A.; Thuwajit, C. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer 2014, 14, 955. [Google Scholar] [CrossRef] [PubMed]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of fak-pi3k/akt pathway. Cell Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; Paige, A.J.; Taylor, K.J.; Ward, C.; Kuske, B.; Zhang, J.; Sun, M.; Janczar, S.; Harrison, D.J.; Muir, M.; et al. Wwox gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin alpha3. Cancer Res. 2009, 69, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Han, X.; Jin, C.; Tian, W.; Yu, W.; Ding, D.; Cheng, L.; Huang, B.; Jiang, H.; Lin, B. Sox2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: New molecular leads for therapeutic intervention. Omics 2013, 17, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; Joffe, E.B.; Simian, M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through beta1 integrin. Breast Cancer Res. Treat. 2012, 133, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, M.; Yang, L.; Tu, G.; Zhu, Q.; Chen, M.; Cheng, H.; Luo, H.; Fu, W.; Li, Z.; et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: A new role for g protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and beta1-integrin signaling pathway in tumor cells. Breast Cancer Res. BCR 2015, 17, 69. [Google Scholar] [PubMed]
- Tomasini-Johansson, B.R.; Kaufman, N.R.; Ensenberger, M.G.; Ozeri, V.; Hanski, E.; Mosher, D.F. A 49-residue peptide from adhesin f1 of streptococcus pyogenes inhibits fibronectin matrix assembly. J. Biol. Chem. 2001, 276, 23430–23439. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T. Extracellular matrix components in breast cancer progression and metastasis. Breast 2013, 22 (Suppl. 2), S66–S72. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V. Anti-adhesion cancer therapy. Cancer Metastasis Rev. 1998, 17, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Varner, J.A. Adhesion molecules in cancer: The role of integrins. Curr. Opin. Cell Biol. 1993, 5, 812–818. [Google Scholar] [CrossRef]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lei, Q.; Guan, K.L. Mst out and hcc in. Cancer Cell 2009, 16, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Bommert, K.; Bargou, R.C.; Stuhmer, T. Signalling and survival pathways in multiple myeloma. Eur. J. Cancer 2006, 42, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Jain, S.; Stuhmer, T.; Andrulis, M.; Ungethum, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Kramer, D.; et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, X.; Chen, B.; Wang, B.; Zhao, Y.; Zhuang, Y.; Shen, H.; Zhang, Z.; Dai, J. A collagen-binding egfr single-chain fv antibody fragment for the targeted cancer therapy. J. Control. Release 2015, 209, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Hascall, V.C.; Atanelishvili, I.; Moreno Rodriguez, R.; Markwald, R.R.; Ghatak, S. Utilization of glycosaminoglycans/proteoglycans as carriers for targeted therapy delivery. Int. J. Cell Biol. 2015, 2015, 537560. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Kouvidi, K.; Kavasi, R.M.; Berdiaki, A.; Tzanakakis, G.N. Hyaluronan/hyaladherins—A promising axis for targeted drug delivery in cancer. Curr. Drug Deliv. 2016, 13, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Cheng, X.B.; Kohi, S.; Koga, A.; Hirata, K. Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm. Sin. B 2016, 6, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Yata, T.; Lee, E.L.; Suwan, K.; Syed, N.; Asavarut, P.; Hajitou, A. Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol. Cancer 2015, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Cun, X.; Chen, J.; Ruan, S.; Zhang, L.; Wan, J.; He, Q.; Gao, H. A novel strategy through combining irgd peptide with tumor-microenvironment-responsive and multistage nanoparticles for deep tumor penetration. ACS Appl. Mater. Interfaces 2015, 7, 27458–27466. [Google Scholar] [CrossRef] [PubMed]
- Cun, X.; Ruan, S.; Chen, J.; Zhang, L.; Li, J.; He, Q.; Gao, H. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomater. 2016, 31, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.V.; Shamay, Y.; Shah, J.; Roxbury, D.; Paknejad, N.; Heller, D.A. Photoluminescent carbon nanotubes interrogate the permeability of multicellular tumor spheroids. Carbon 2016, 97, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, S.; Chelvam, V.; Low, P.S. Comparison of nanoparticle penetration into solid tumors and sites of inflammation: Studies using targeted and nontargeted liposomes. Nanomedicine 2015, 10, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Villegas, M.R.; Baeza, A.; Vallet-Regi, M. Hybrid collagenase nanocapsules for enhanced nanocarrier penetration in tumoral tissues. ACS Appl. Mater. Interfaces 2015, 7, 24075–24081. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popovic, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Jiang, T.; Shen, S.; She, X.; Tuo, Y.; Hu, Y.; Pang, Z.; Jiang, X. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016, 103, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Veale, R.B.; Thornley, A.L. Increased single class low affinity EGF receptors expressed by human oesophageal squamous carcinoma cell lines. S. Afr. J. Sci. 1989, 85, 375–379. [Google Scholar]
- Shimada, Y.; Imamura, M.; Wagata, T.; Yamaguchi, N.; Tobe, T. Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992, 69, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Kaschula, C.H.; Hunter, R.; Stellenboom, N.; Caira, M.R.; Winks, S.; Ogunleye, T.; Richards, P.; Cotton, J.; Zilbeyaz, K.; Wang, Y.; et al. Structure-activity studies on the anti-proliferation activity of ajoene analogues in whco1 oesophageal cancer cells. Eur. J. Med. Chem. 2012, 50, 236–254. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Vogelsang, M.; Parker, M.I. Wnt/beta-catenin and MEK-ERK signaling are required for fibroblast-derived extracellular matrix-mediated endoderm differentiation of embryonic stem cells. Stem Cell Rev. 2015, 11, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Leaner, V.D.; Parker, M.I. Absence of feedback regulation in the synthesis of COL1A1. Life Sci. 2014, 103, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biopsy Number | Histology | Sex | Age | Tumor Differentiation (Grade) | Tumor Site ICD-10 | Invasive or Infiltrating |
|---|---|---|---|---|---|---|
| 543 | ESCC | M | 55 | ND | C15.4 | Infiltrating |
| 547 | ESCC | F | 30 | Moderate | C15.5 | Invasive |
| 551 | ESCC | M | 47 | Moderate | C15.5 | Invasive |
| 556 | ESCC | F | 54 | Moderate | C15.4 | Invasive |
| 561 | ESCC | M | 58 | Moderate | C15.9 | Keratinizing |
| 563 | ESCC | M | 52 | Moderate | C15.5 | Infiltrating |
| 569 | ESCC | F | 79 | Poor | C15.4 | Invasive |
| 571 | ESCC | F | 48 | Moderate | C15.3 | Keratinizing |
| 573 | ESCC | F | 41 | ND | C15.3 | Infiltrating |
| 591 | ESCC | M | 47 | Moderate | C15.4 | Invasive |
| 596 | ESCC | F | 67 | Moderate | C15.4 | Invasive |
| 601 | ESCC | M | 59 | ND | C15.4 | Infiltrating |
| 607 | ESCC | F | 48 | Moderate | C15.4 | ND |
| 613 | ESCC | M | 54 | Moderate | C15.9 | Invasive |
| 618 | ESCC | F | 60 | Moderate | C15.4 | Keratinizing |
| 619 | ESCC | M | 57 | Moderate | C15.4 | Infiltrating |
| 621 | ESCC | F | 64 | Moderate | C15.4 | Invasive |
| 622 | ESCC | F | 83 | ND | C15.4 | Infiltrating |
| 627 | ESCC | M | 52 | Moderate | ND | ND |
| 634 | ESCC | F | 57 | Moderate | C15.4 | Keratinizing |
| 635 | ESCC | M | 57 | Moderate | C15.4 | Keratinizing |
| Glycoproteins | Collagens | ECM Regulators | ECM Affiliated Proteins | Secreted Factors | Proteoglycans |
|---|---|---|---|---|---|
| Gene Name | |||||
| FN1 | COL1A1 | TGM2 | LGALS1 | S100A13 | HSPG2 |
| LAMA3 | COL1A2 | HTRA1 | FREM2 | EGFL7 | BGN |
| LAMA5 | COL6A3 | CSTB | ANXA2 | IGF2 | DCN |
| FBN1 | COL3A1 | LOXL2 | FREM1 | S100A11 | LUM |
| TGFB1 | COL12A1 | LOXL1 | ANXA6 | S100A6 | ASPN |
| TNC | COL6A1 | SERPINH1 | ANXA5 | S100A13 | OGN |
| EMILIN1 | COL4A2 | CTSB | COLC12 | CXCL12 | PRELP |
| LAMC1 | COL6A2 | LOX | CLEC3B | CCL25 | VCAN |
| LAMB2 | COL4A5 | ITH5 | LGALS3 | PF4 | |
| FBLN2 | COL4A4 | ADAM10 | LGALS8 | FGF2 | |
| LAMA2 | COL5A2 | ADAMTSL1 | SEMA3C | INSL5 | |
| TNXB | COL7A1 | PLG | CLEC14A | ANGPTL2 | |
| POSTN | COL11A1 | PZP | ANXA9 | S100A9 | |
| THBS1 | COL4A1 | CTSK | ANXA1 | ||
| FBN2 | COL5A1 | ADAMTSL5 | PLXDC2 | ||
| FBLN1 | COL5A3 | SERPINA1A | SFTPA1 | ||
| LAMB3 | COL14A1 | SERPINA3K | CSPG4 | ||
| LAMA4 | COL16A1 | PLOD1 | SFTPD | ||
| AGRN | COL18A1 | ||||
| FGB | COL15A1 | ||||
| LAMC2 | |||||
| VWF | |||||
| HMCN1 | |||||
| LTBP4 | |||||
| Drug | Plastic | tfd-ECM | cd-ECM | combi-ECM |
|---|---|---|---|---|
| Cisplatin (IC50 ± S.D. (µM)) | 18.5 ± 6.4 | 23.8 ± 3.2 | 22.4 ± 4.5 | 25.7 ± 3.2 |
| 5-FU (IC50 ± S.D. (µM)) | 14.1 ± 3.8 | 19.1 ± 2.6 | 20.6 ± 2.2 | 21.9 ± 1.8 |
| Epirubicin (IC50 ± S.D. (µM)) | 12.8 ± 2.3 | 17.3 ± 4.5 | 18.5 ± 1.9 | 27.8 ± 5.3 |
| Plastic | tfd-ECM | cd-ECM | combi-ECM | |
|---|---|---|---|---|
| No Drug (hours) | 33.6 ± 3.3 | 38.6 ± 5.7 | 37.1 ± 4.2 | 36.8 ± 4.5 |
| Cisplatin (hours) | 55.3 ± 9.4 | 39.5 ± 4.3 | 36.9 ± 3.8 | 36.7 ± 5.8 |
| 5-FU (hours) | 56.2 ± 5.1 | 39.5 ± 3.6 | 32.6 ± 4.6 | 31.9 ± 3.8 |
| Epirubicin (hours) | 58.3 ± 2.5 | 34.7 ± 3.5 | 30.7 ± 4.9 | 32.1 ± 3.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102861
Senthebane DA, Jonker T, Rowe A, Thomford NE, Munro D, Dandara C, Wonkam A, Govender D, Calder B, Soares NC, et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. International Journal of Molecular Sciences. 2018; 19(10):2861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102861
Chicago/Turabian StyleSenthebane, Dimakatso Alice, Tina Jonker, Arielle Rowe, Nicholas Ekow Thomford, Daniella Munro, Collet Dandara, Ambroise Wonkam, Dhirendra Govender, Bridget Calder, Nelson C. Soares, and et al. 2018. "The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices" International Journal of Molecular Sciences 19, no. 10: 2861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102861