Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway

 , , and
, , and 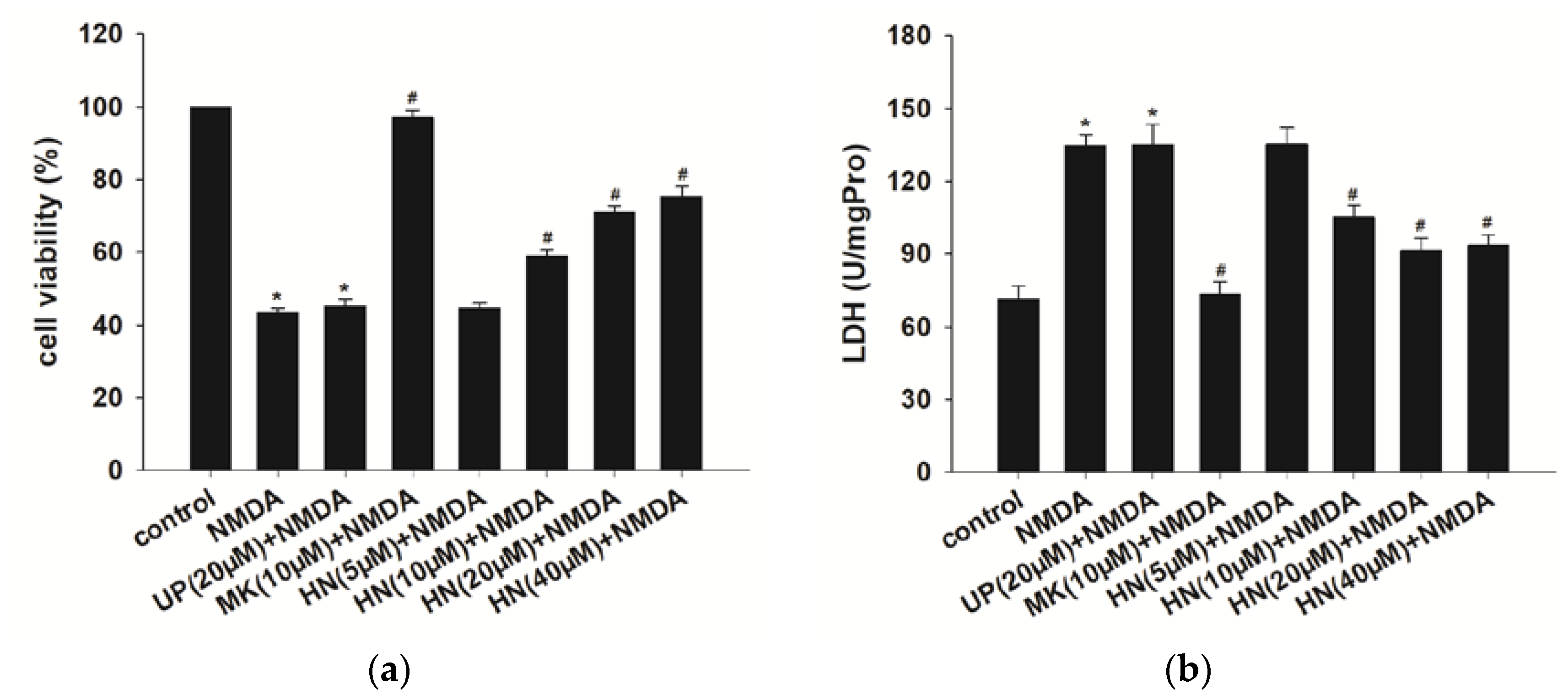{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
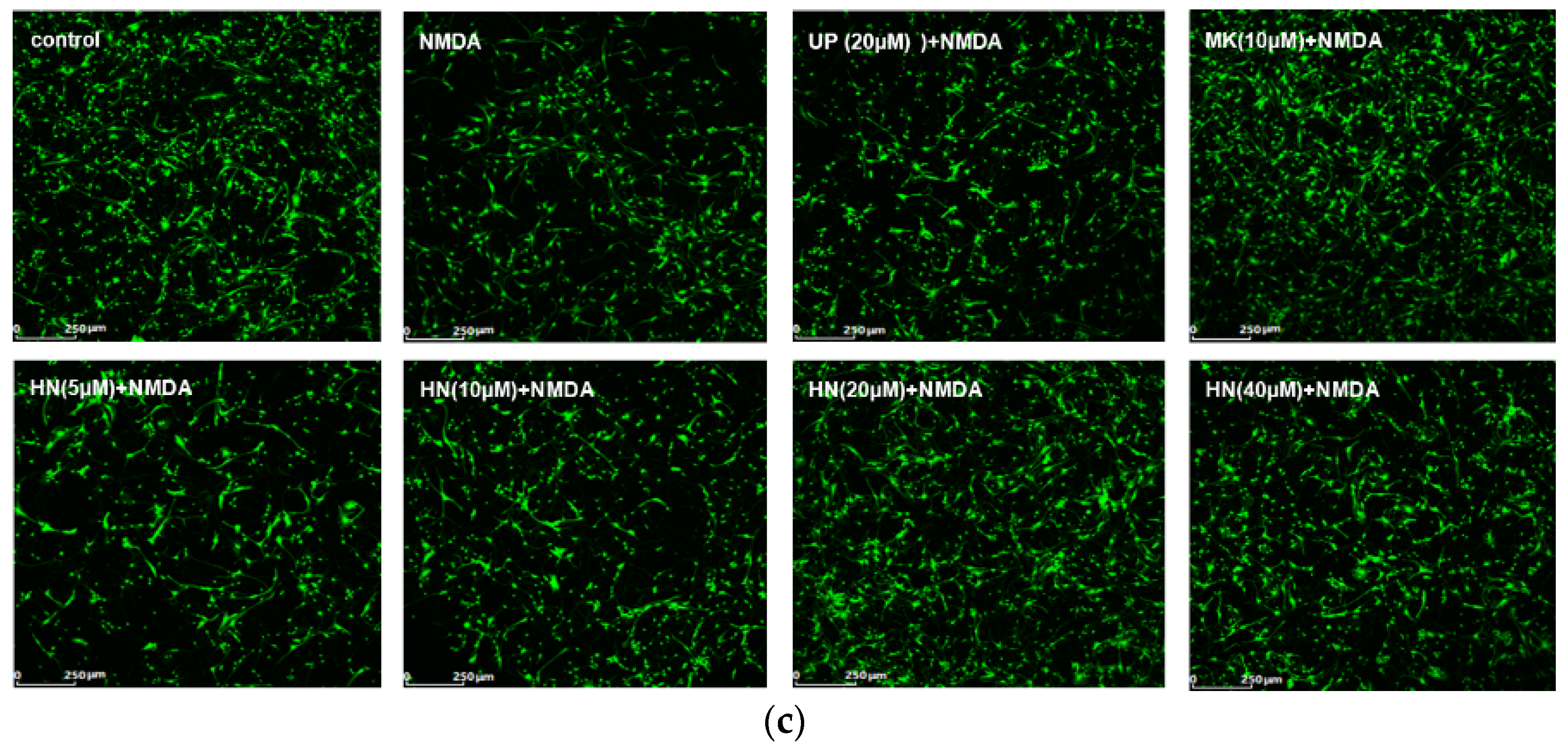
2.1. Humanin (HN) Prevents from NMDA-Induced Neuronal Insults
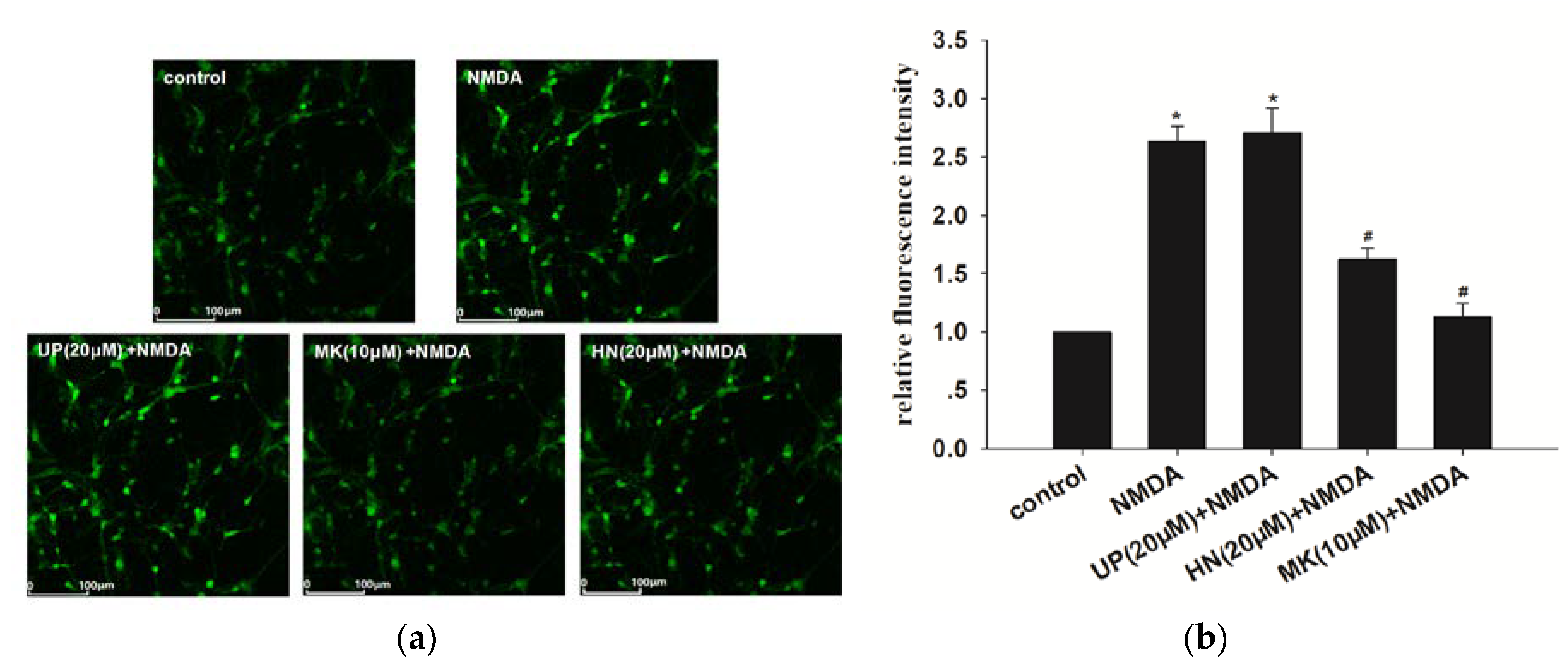
2.2. Humanin (HN) Attenuated NMDA-Induced Elevation of Intracellular Calcium
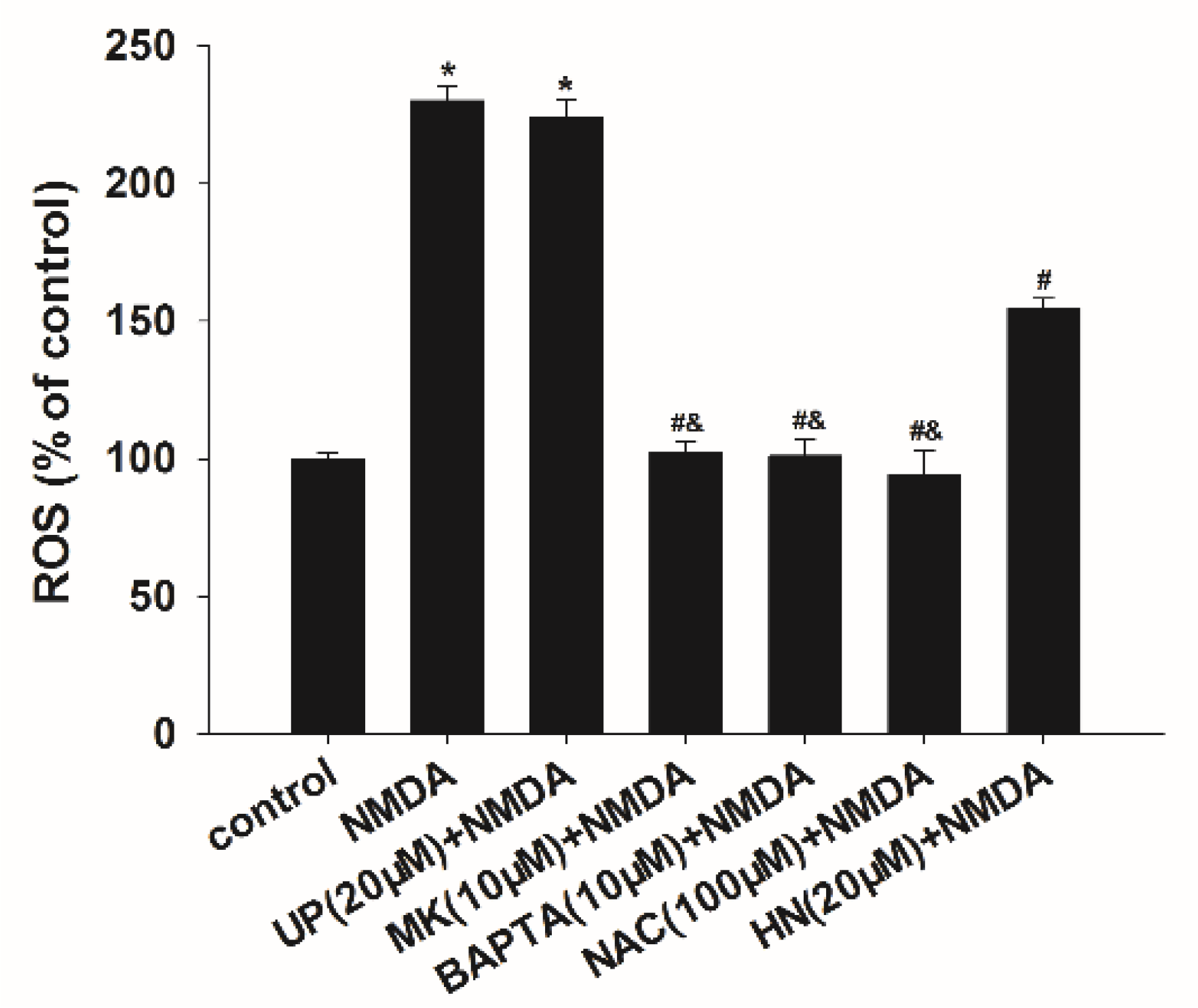
2.3. Decrease in Reactive Oxygen Species Levels by HN, BAPTA-AM or NAC after NMDA Exposure
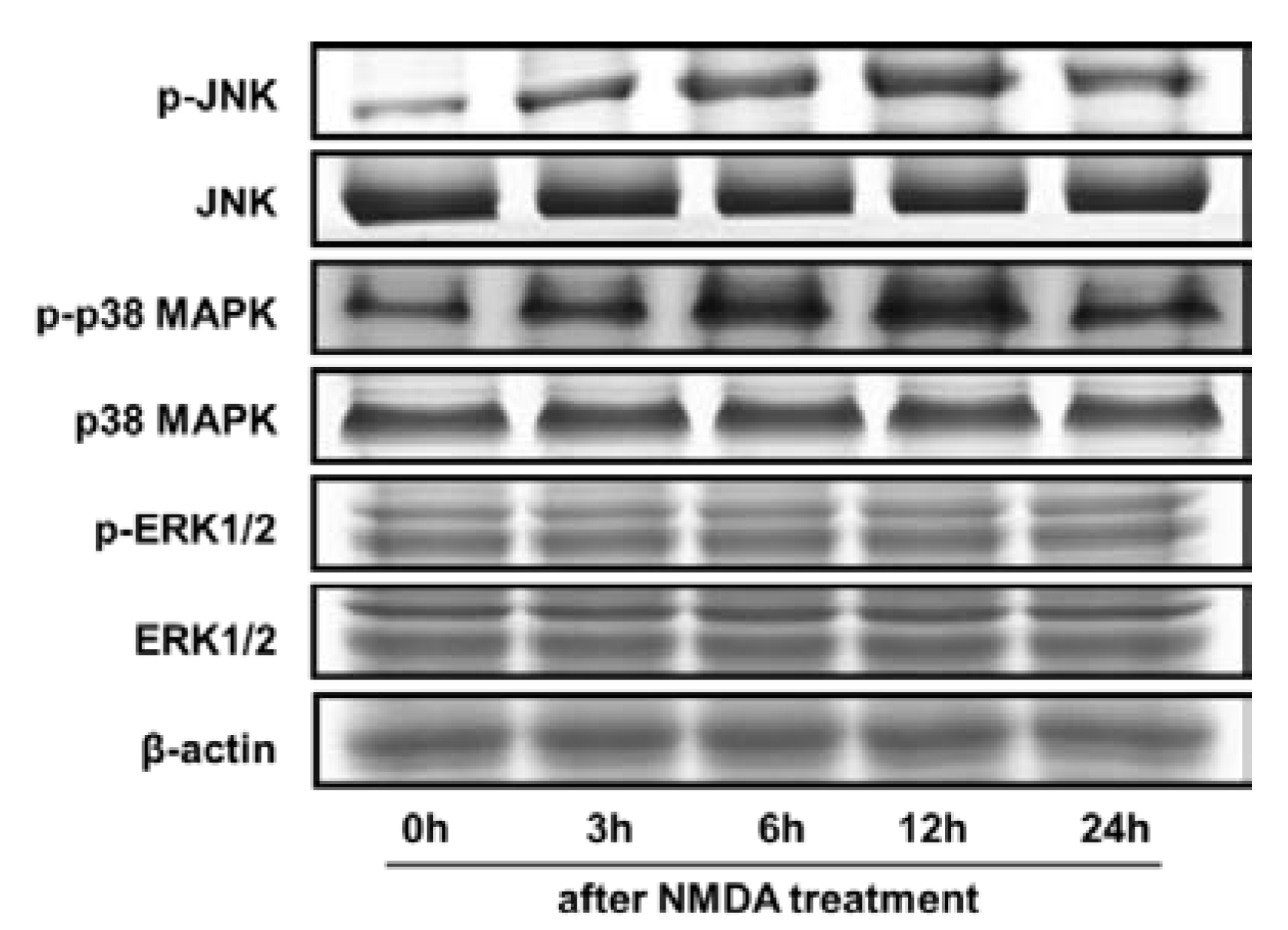
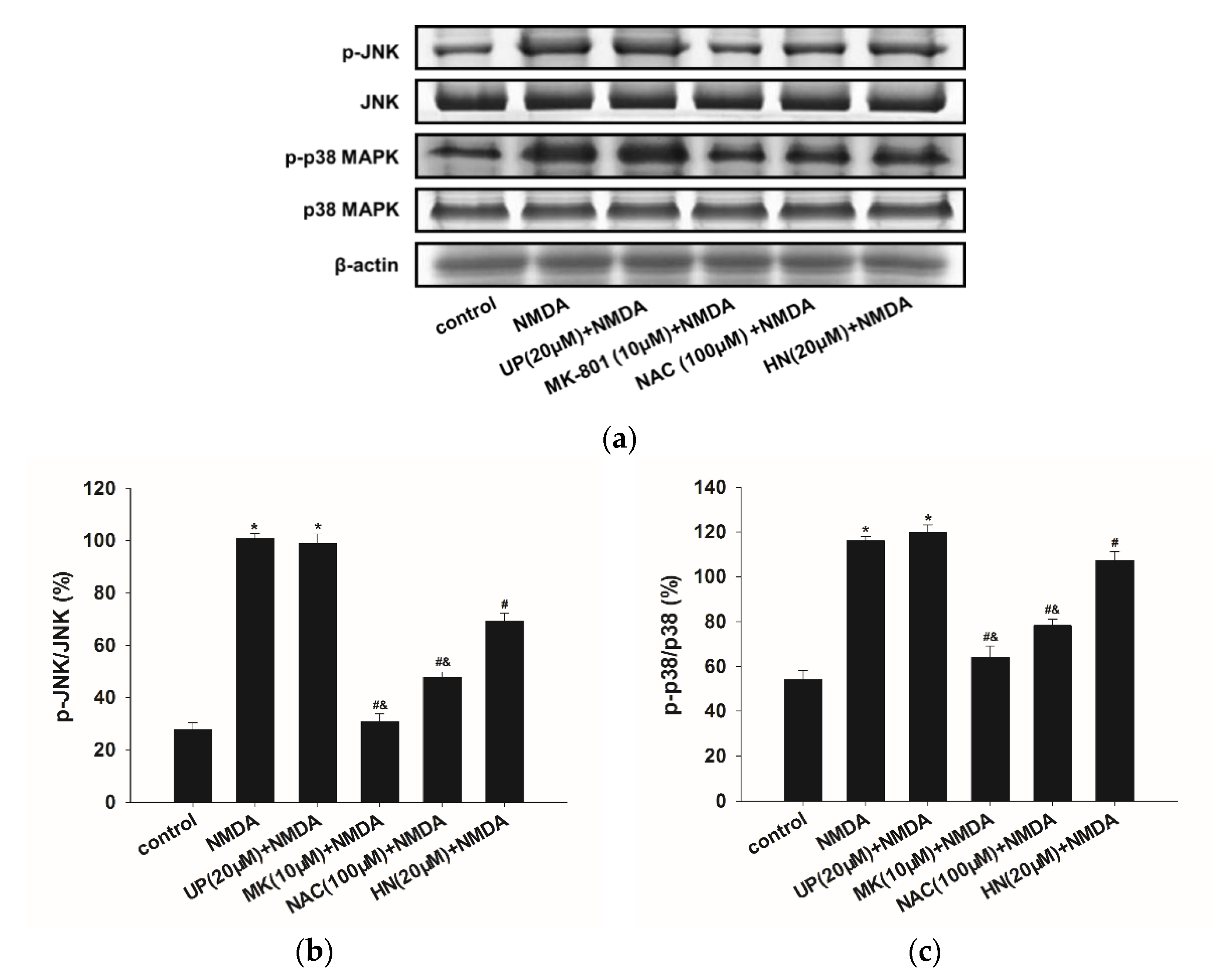
2.4. NMDA-Induced MAPKs Activation were Attenuated by HN or NAC
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal and Cell Culture
4.3. Treatments
4.4. Cell Viability Assay
4.5. Lactate Dehydrogenase (LDH) Assay
4.6. Calcein-AM Assay
4.7. Confocal Calcium Imaging/Measurement
4.8. Measurement of Reactive Oxygen Species (ROS)
4.9. Western Blot Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| HN | Humanin |
| UP | unrelated peptide |
| NMDA | N-methyl-d-aspartate |
| MK-801 | 5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate |
| MAPKs | Mitogen- activated protein kinases |
| ERK1/2 | extracellar signal-regulated protein kinase |
| JNK | c-Jun N-terminal kinase |
| BAPTA-AM | 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid |
| LDH | lactate dehydrogenase |
| AD | Alzheimer’s disease |
| ROS | reactive oxygen species |
| NAC | N-Acetyl-l-cysteine |
| H2DCFDA | 2’,7’-dichlorodihydrofluorescein diacetate |
| FPRL | formyl peptide-receptor-like |
| CNTFR | ciliary neurotrophic factor receptor α |
References
- Greengard, P. The neurobiology of slow synaptic transmission. Science 2001, 294, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Gonda, X. Basic pharmacology of NMDA receptors. Curr. Pharm. Des. 2012, 18, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; Bhat, J.A.; Satti, N.K.; Sharma, P.R.; Hamid, A.; Ahmad, M. Withanone, an Active Constituent from Withania somnifera, Affords Protection Against NMDA-Induced Excitotoxicity in Neuron-Like Cells. Mol. Neurobiol. 2017, 54, 5061–5073. [Google Scholar] [CrossRef] [PubMed]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Gu, Z.; Zhang, G.; Jing, G. Diphosphorylation and involvement of extracellular signal-regulated kinases (ERK1/2) in glutamate-induced apoptotic-like death in cultured rat cortical neurons. Brain Res. 2000, 857, 71–77. [Google Scholar] [CrossRef]
- Pi, R.; Li, W.; Lee, N.T.; Chan, H.H.; Pu, Y.; Chan, L.N.; Sucher, N.J.; Chang, D.C.; Li, M.; Han, Y. Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J. Neurochem. 2004, 91, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.W.; Park, K.Y.; Kim, H.; Han, P.L.; Kim, Y.U.; Gwag, B.J.; Choi, E.J. Ca2+-mediated activation of c-Jun N-terminal kinase and nuclear factor kappa B by NMDA in cortical cell cultures. J. Neurochem. 1998, 71, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Ali, C.; Docagne, F.; Plawinski, L.; MacKenzie, E.T.; Vivien, D.; Buisson, A. Neuroprotection mediated by glial cell line-derived neurotrophic factor: Involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J. Neurosci. 2001, 21, 3024–3033. [Google Scholar] [CrossRef] [PubMed]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.M.; Geddes-Klein, D.M.; Hockenberry, A.; Scarsella, D.; Mesfin, M.N.; Singh, P.; Patel, T.P.; Meaney, D.F. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem. Int. 2012, 60, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Gladding, C.M.; Wang, L.; Zhang, L.Y.; Kaufman, A.M.; Milnerwood, A.J.; Raymond, L.A. P38 MAPK is involved in enhanced NMDA receptor-dependent excitotoxicity in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 2012, 45, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; Satti, N.K.; Dutt, P.; Hamid, A.; Ahmad, M. Attenuation of Glutamate-Induced Excitotoxicity by Withanolide-A in Neuron-Like Cells: Role for PI3K/Akt/MAPK Signaling Pathway. Mol. Neurobiol. 2017, 55, 2725–2739. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Sun, P.; Qin, H.P.; Si, P.P.; Sun, X.F.; Zhang, C. Involvement of MAPK pathways in NMDA-induced apoptosis of rat cortical neurons. Sheng Li Xue Bao 2012, 64, 609–616. [Google Scholar] [PubMed]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer‘s disease genes and Abeta. Proc. Natl. Acad Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ito, Y.; Niikura, T.; Shao, Z.; Hata, M.; Oyama, F.; Nishimoto, I. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem. Biophys. Res. Commun. 2001, 283, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Tajima, H.; Niikura, T.; Hashimoto, Y.; Ito, Y.; Kita, Y.; Terashita, K.; Yamazaki, K.; Koto, A.; Aiso, S.; Nishimoto, I. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer‘s disease-related insults. Neurosci. Lett. 2002, 324, 227–231. [Google Scholar] [CrossRef]
- Zapala, B.; Kaczynski, L.; Kiec-Wilk, B.; Staszel, T.; Knapp, A.; Thoresen, G.H.; Wybranska, I.; Dembinska-Kiec, A. Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties. Pharmacol. Rep. 2010, 62, 767–777. [Google Scholar] [CrossRef]
- Kumfu, S.; Charununtakorn, S.T.; Jaiwongkam, T.; Chattipakorn, N.; Chattipakorn, S.C. Humanin Exerts Neuroprotection during Cardiac Ischemia-Reperfusion Injury. J. Alzheimers Dis. 2018, 61, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.L.; Zhang, Y.H.; Li, J.Z.; Song, T.; Liu, X.M.; Wang, H.; Zhang, C.; Ma, G.L.; Zhang, H.; Li, K. Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction. Drug Des. Devel. Ther. 2017, 11, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.P.; Bilousova, T.; Spilman, P.; Vadivel, K.; Bai, D.; Elias, C.J.; Evseenko, D.; John, V. A Small Molecule Mimetic of the Humanin Peptide as a Candidate for Modulating NMDA-Induced Neurotoxicity. ACS Chem. Neurosci. 2018, 9, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, D.; Li, L.; Zhao, W.; Zhang, C. Protective effects of humanin on okadaic Acid-induced neurotoxicities in cultured cortical neurons. Neurochem. Res. 2014, 39, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Niikura, T.; Hashimoto, Y.; Tajima, H.; Nishimoto, I. Death and survival of neuronal cells exposed to Alzheimer‘s insults. J. Neurosci. Res. 2002, 70, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Kumfu, S.; Charununtakorn, S.T.; Jaiwongkam, T.; Chattipakorn, N.; Chattipakorn, S.C. Humanin prevents brain mitochondrial dysfunction in a cardiac ischaemia-reperfusion injury model. Exp. Physiol. 2016, 101, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.E.; Cui, L.; Gong, Z.; Su, K.; Muzumdar, R. A humanin analog decreases oxidative stress and preserves mitochondrial integrity in cardiac myoblasts. Biochem. Biophys. Res. Commun. 2013, 440, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, R.; Charlton, M.P.; Hafner, M.; Tymianski, M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J. Neurochem. 1998, 71, 2349–2364. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.L.; Li, J.Z.; Feng, Z.B.; Ma, G.L.; Gong, L.; Li, C.L.; Zhang, C.; Li, K. Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity not by NMDA receptor. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Habata, Y.; Hosoya, M.; Nishi, K.; Fujii, R.; Kobayashi, M.; Hinuma, S. N-Formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun. 2004, 324, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Iribarren, P.; Zhou, Y.; Gong, W.; Zhang, N.; Yu, Z.X.; Le, Y.; Cui, Y.; Wang, J.M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004, 172, 7078–7085. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, E.M.; Sanz-Blasco, S.; Karachitos, A.; Bandez, M.J.; Fernandez-Gomez, F.J.; Perez-Alvarez, S.; de Mera, R.M.; Jordan, M.J.; Aguirre, N.; Galindo, M.F.; et al. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem. Pharmacol. 2010, 79, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Centeno, C.; Repici, M.; Chatton, J.Y.; Riederer, B.M.; Bonny, C.; Nicod, P.; Price, M.; Clarke, P.G.; Papa, S.; Franzoso, G.; et al. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Badaut, J.; Thevenet, J.; Granziera, C.; Regli, L.; Maurer, F.; Bonny, C.; Bogousslavsky, J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004, 35, 1738–1743. [Google Scholar] [CrossRef] [PubMed]
- Legos, J.J.; Erhardt, J.A.; White, R.F.; Lenhard, S.C.; Chandra, S.; Parsons, A.A.; Tuma, R.F.; Barone, F.C. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 2001, 892, 70–77. [Google Scholar] [CrossRef]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999, 96, 12866–12869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaper, S.D.; Facci, L.; Strijbos, P.J. Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann. N. Y. Acad. Sci. 2001, 939, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, S.W.; Bottum, K.M.; Krager, S.L.; Tischkau, S.A. ERK/MAPK is essential for endogenous neuroprotection in SCN2.2 cells. PLoS ONE 2011, 6, e23493. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Schnellmann, R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Kelkel, M.; Cerella, C.; Mack, F.; Schneider, T.; Jacob, C.; Schumacher, M.; Dicato, M.; Diederich, M. ROS-independent JNK activation and multisite phosphorylation of Bcl-2 link diallyl tetrasulfide-induced mitotic arrest to apoptosis. Carcinogenesis 2012, 33, 2162–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.K.; Kim, E.H.; Choi, M.A.; Park, J.M.; Kim, D.H.; Surh, Y.J. Diallyl trisulfide induces apoptosis in human breast cancer cells through ROS-mediated activation of JNK and AP-1. Biochem. Pharmacol. 2012, 84, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.B.; Choi, J.; Baek, S.J.; Lee, S.H. Reactive oxygen species mediate tolfenamic acid-induced apoptosis in human colorectal cancer cells. Arch. Biochem. Biophys. 2013, 537, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, F.; Li, F.; Fu, H.; Wang, J.; Zhang, S.; Zhao, J.; Yin, D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem. Biophys. Res. Commun. 2015, 463, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; Ma, C.; Qiao, J.; Zhang, C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat‘s cultured cortical neurons. Neurosci. Lett. 2008, 447, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Lin, L.; Lin, F.; Li, T.; Du, H.; Chen, R.; Zheng, W.; Liu, N. Effects of bone marrow-derived mesenchymal stem cells on the axonal outgrowth through activation of PI3K/AKT signaling in primary cortical neurons followed oxygen-glucose deprivation injury. PLoS ONE 2013, 8, e78514. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, M.C.; Yard, M.; Jackson, J.; Lahiri, D.K.; Kubek, M.J. An analog of thyrotropin-releasing hormone (TRH) is neuroprotective against glutamate-induced toxicity in fetal rat hippocampal neurons in vitro. Brain Res. 2007, 1128, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhang, H.; Wu, J.; Yin, L.; Yan, L.-J.; Zhang, C. Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102982
Yang X, Zhang H, Wu J, Yin L, Yan L-J, Zhang C. Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway. International Journal of Molecular Sciences. 2018; 19(10):2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102982
Chicago/Turabian StyleYang, Xiaorong, Hongmei Zhang, Jinzi Wu, Litian Yin, Liang-Jun Yan, and Ce Zhang. 2018. "Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway" International Journal of Molecular Sciences 19, no. 10: 2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102982