The Role of the TGFβ Receptor Signaling Pathway in Adult Beta Cell Proliferation


1
Division of Pediatric Surgery, Department of Surgery, Children′s Hospital of Pittsburgh, University of Pittsburgh School of Medicine, 4401 Penn Ave, Pittsburgh, PA 15224, USA
2
The Warren Alpert Medical School of Brown University, 222 Richmond Street, Providence, RI 02903, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(10), 3136; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103136
Submission received: 26 July 2018
/
Revised: 9 October 2018
/
Accepted: 10 October 2018
/
Published: 12 October 2018
(This article belongs to the Special Issue TGF-Beta Super Family Signaling)
{kind=link}
Abstract
:Diabetes is a global epidemic and affects millions of individuals in the United States. Devising novel treatments for diabetes continues to be a great medical challenge. Postnatal beta cell growth or compensation is largely attributed to beta cell proliferation, which declines continuously with age. To boost beta cell proliferation to regenerate an adequate functional mass, there is a need to understand the signaling pathways that regulate beta cell proliferation for creating practical strategies to promote the process. Transforming growth factor β (TGFβ) belongs to a signaling superfamily that governs pancreatic development and the regeneration of beta cells after pancreatic diseases. TGFβ exerts its functions by activation of downstream Smad proteins and through its crosstalk with other pathways. Accumulating data demonstrate that the TGFβ receptor signaling pathway also participates in the control of beta cell proliferation. This review details the role of the TGFβ receptor signaling pathway in beta cell proliferation physiologically and in the pathogenesis of diabetes.
1. Introduction
Insulin is a key regulator of glucose homeostasis, and is produced exclusively by pancreatic beta cells. Inadequate secretion or action of insulin causes diabetes, a metabolic disease characterized by high blood glucose, persistence of which will lead to the classical symptoms of polyuria, polydipsia and polyphagia [1]. There are two major types of diabetes. While Type 1 diabetes (T1D) primarily results from the body’s failure to produce insulin due to autoimmunity to insulin-producing beta cells, Type 2 diabetes (T2D) primarily results from insulin resistance, a condition in which cells fail to use insulin properly; with time, these patients can develop an absolute insulin deficiency. In the United States, T1D and T2D affect approximately 30 million individuals [1]. The WHO estimates that there are about 347 million individuals who have diabetes across the globe. T1D and T2D are both associated with a deficiency of beta cells, although they are different diseases. Beta cell replication is viewed as the basic mechanism underlying beta cell generation and maintenance. However, human beta cell proliferation declines rapidly by 1 year of age [2,3]. Thus, diabetes patients will benefit from beta cell regeneration and replacement strategies.
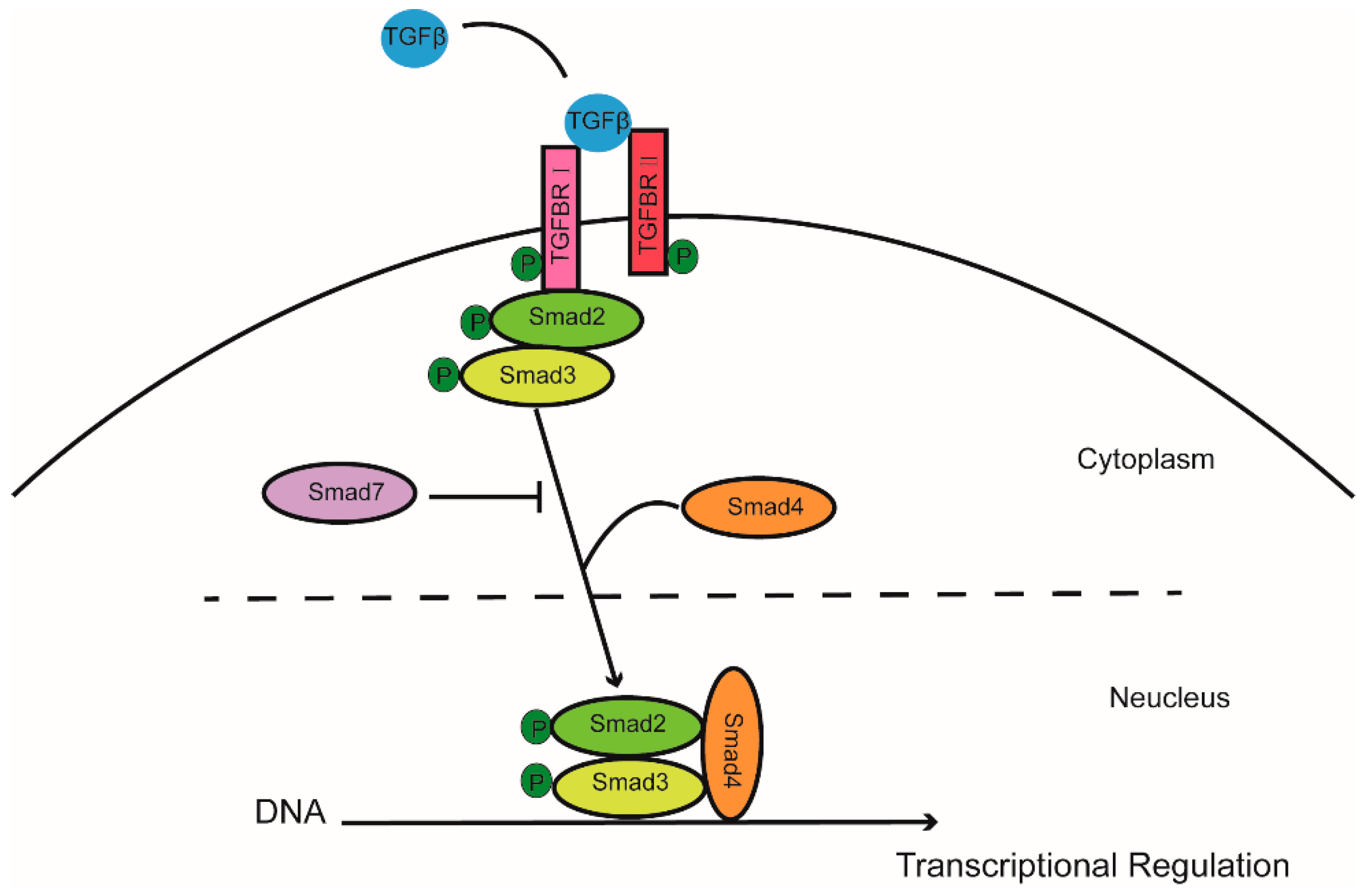
Transforming growth factor β (TGFβ) belongs to a signaling superfamily which acts on plasma receptors to induce multiple biological effects, including development, immune response, and cell growth. In short, TGFβ binds to type II receptors on the cell membrane, recruiting type I receptors which then phosphorylate Smad2 and Smad3 (R-Smad). After forming a complex with Smad4, they translocate from the cytoplasm to the nucleus and regulate downstream gene expression. Activation of the TGFβ receptor signaling pathway may be inhibited by Smad6 and Smad7 [4] (Figure 1). TGFβ receptor signaling pathway plays roles in many cellular aspects through canonical SMAD pathway or noncanonical pathways, including mitogen-activated protein kinase (MAPK) pathways and the phosphatidylinositol 3′-kinase (PI3K)-protein kinase B (AKT) pathway. For example, Hamidi et al. showed that TGFβ activated PI3K in a tumor necrosis factor receptor-associated factor 6 (TRAF6)-dependent manner [5]. The TGFβ receptor signaling pathway has effects on beta cell proliferation and phenotype in mouse models. We have previously shown that Smad7, a potent suppressor of TGFβ receptor signaling, plays an essential role in maintenance of beta cell mass and in postnatal beta cell proliferation under certain circumstances [6,7,8]. In this review, we focus on the regulatory role of the TGFβ receptor signaling pathway in beta cell proliferation.
2. Beta Cell Proliferation and Cell Cycle Progression
The adult human pancreas contains about 1–2 g of beta cell mass, which plays an important role in glucose homeostasis [9]. During the embryonic period, human beta cells are primarily derived from precursor cells, after which proliferation of beta cells speeds up to generate functional beta cell mass. Significant increases in beta cell mass was observed to peak within the first 2 years of life, and then to rapidly decline in early childhood [10,11]. In the first year after birth, 1–3% of beta cells are in the active cell cycle, whereas almost 50% of cells from other organs could proliferate actively. In adults, human beta cell proliferation is very low, as no proliferation of beta cells was detected in 18 adult human pancreas samples based on staining for Ki-67, a specific cell proliferation marker that labels cells in an active cell cycle [12]. However, a recent study showed that the poor quality of Ki-67 staining in human tissue may result in underestimation of the real cell proliferation [13]. Hence, improvement of the technology or seeking alterative cell proliferation markers may be needed to determine and quantify beta cell growth in human. Although rodent beta cells exhibit better proliferation potential than human beta cells, generally, it is also very low [14].
Cell proliferation is first regulated at the G1/S cell phase, which controls the entry into the cell cycle. Given the low proliferation index of adult beta cells, it seems that cell cycle progression of beta cells is regulated by molecules which regulate G1/S transition [11,15,16]. Human adult beta cells contain not only cyclin-dependent kinases (CDKs) and transcription factors such as E2F that direct entry into the cell cycle, but also cyclin-dependent kinases inhibitors (CDKIs), as well as pocket proteins, including pRb, p107 and p130, which prevent cell cycle progression [17,18,19]. Replication of adult human beta cells cannot occur spontaneously or be easily stimulated by mitogen stimulation, largely due to the presence of CDKIs, which play a critical inhibitory role in cell cycle control. Overexpression of CDKs or cyclins in human islets transplanted into diabetic mice induced replication of adult beta cells and reverse diabetes effectively [20]. Blocking a CDKI, p57KIP2, by shRNA promoted entry into the G1/S phase. Inactivation of other CDKIs (such as p18INK4c, p21CIP1 and p27KIP1) induces familial syndromes of endocrine-cell hyperplasia [21,22]. In rodent beta cells, it was also observed that some cyclins, such as cyclinD2 and CDK6, were located in cytoplasm, rather than in nucleus in other cell types [19,23]. Similar result was reported in human beta cell [23]. Hence, the improper location of cyclins may also retrain beta cell proliferation in both rodents and humans. The proper balance of CDKs and CDKIs is likely coordinated to regulate the replication of adult beta cells.
3. TGFβ in Beta Cell Proliferation and Function
Past studies have shown that the TGFβ receptor signaling pathway participates in pancreatic development, as well as pancreatic disease like pancreatitis and pancreatic carcinoma [24,25,26,27]. TGFβ receptor signaling promotes endocrine cell differentiation and maturation and inhibits acinar cell growth during embryogenesis [28,29,30]. Our own observations have shown that beta cell proliferation occurs following pancreatic duct ligation (PDL), and that this proliferation depends on infiltrating macrophages [6]. Macrophages not only release TGFβ1 to upregulate Smad7, but also secrete Epidermal Growth Factor (EGF) to activate the EGF pathway, which inhibits the nuclear translocation of the phosphorylated SMAD complex. This inflammation-induced beta cell proliferation was associated with increased levels of Cyclin D1, Cyclin D2 and nuclear exclusion of p27 regulated by Smad7 [6]. TGFβ receptor signaling appears to be not essential for beta cell proliferation after partial pancreatectomy (PPx), an increased-workload-induced beta cell proliferation model [31], although inhibition of the TGFβ receptor signaling pathway may slightly increase beta cell proliferation and induce beta cell mass 1 week after PPx [32]. These results suggest that TGFβ receptor signaling is a central regulator of beta cell proliferation and homeostasis. Smad7, as an antagonist of the TGFβ receptor signaling pathway, appears to be a direct trigger of beta cell proliferation [6]. In adult islets, TGFβ1, Smad2 and Smad3 are expressed continuously while Smad7 is not detectable in insulin, glucagon and somatostatin cells. Smad7 expression re-emerged in cells after pancreas injury associated with increased beta cell proliferation [6,8]. Interestingly, inhibition of Smad7 expression in beta cells leads to a reduction in beta cell proliferation [8,33].
The TGFβ receptor signaling pathway also plays a key role in beta cell function. Totsuka Y. et al. showed that the insulin secretion increased by cultured islets which was incubated with TGFβ1, and this process was dependent on the activation of Smad2 [34,35,36]. Smad7 also participates in beta cell function and insulin secretion, since overexpression of Smad7 in beta cells results in reversible diabetes in mice [37]. Glucose concentration also influences insulin secretion induced by TGFβ1. TGFβ receptor signaling was shown to be crucial for insulin secretion in 200 mg/dL glucose concentrations without effects on beta cell proliferation, while TGFβ1 no longer induced insulin secretion when blood glucose reached 300 mg/dL [35,38]. These results suggest a central role for the TGFβ receptor signaling pathway in regulating beta cell function.
4. Crosstalk of TGFβ with Other Pathways in Beta Cell Proliferation
Inflammation occurs in both T1D and T2D and plays an important role in cell proliferation [39,40,41,42]. IL-1β, secreted by islets after hyperglycemic stimulation, participates in beta cell functional impairment and apoptosis [43]. IL-1β binds to its receptor (IL-1R) and triggers activation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling pathway, which leads to FAS receptor upregulation and beta cell apoptosis [44,45]. Preventing expression of IL-1β could reverse diminished insulin expression and impaired beta cell function. Transgenic mice that express beta cell-specific NF-κB inhibitor demonstrate resistance to streptozotocin induced diabetes [46,47]. In summary, IL-1β and NF-κB mediate glucotoxic islet inflammation and beta cell apoptosis in diabetes.
TGFβ may also participate in glucotoxic islet inflammation and interacts with the NF-κB signaling pathway. Several studies have revealed that TGFβ family genes, including BMP5 and SMAD7, are upregulated by hyperglycemia. An abnormal reaction of TGFβ signaling leads to beta cell functional impairment and increased blood glucose [43,46,47,48]. TGFβ regulates keratinocyte differentiation and proliferation through interactions between smad2, smad3 and NF-κB submit IκB, kinase α (IKKα) [48]. Smad3 interacts with an NF-κB submit and regulates epithelial cell transcriptional activity and ECM related genes in fibroblasts [49]. It has also been reported that NF-κB suppresses the TGFβ signaling pathway by Smad7 activation. Together, crosstalk between NF-κB and TGFβ mediates numerous essential cell functions in multiple organs. However, the nature of interactions between TGFβ and NF-κB in beta cells remains to be studied.
It has also been reported that TGFβ and Insulin-like Growth Factor 1 Receptor (IGFR) signaling regulates the function of beta cells [50]. TGFβ and IGFR signaling are involved in cell proliferation through interaction with PI3K and R-Smads. FOXO family proteins are at the center of this process. Interaction between FOXO and Smad3/4 complex actives cell cycle inhibitor p21, which prevents neuroepithelial cell proliferation. Nucleus translocation and transcriptional function of FOXO could be inhibited by AKT, a downstream molecule of PI3K signaling pathway [51,52]. Negative regulation of FOXO by insulin receptor/IGFR signaling pathway has been shown to induce expression of Pdx1, MafA, the essential beta cell transcriptional factor [52]. Thus, FOXO appears to be a potential factor to regulate beta cell proliferation through coordinating the crosstalk between TGFβ and IGFR pathway.
It is well known that the EGF signaling pathway affects beta cell proliferation [53,54]. Global EGF receptor deficiency leads to a significant deficit in beta cell proliferation in the juvenile period, when the greatest proliferation of beta cells occurs. Similar results have been found in high fat diet and pregnancy mice. It was reported that TGFβ upregulates EGF expression via activation of MAPK and AKT [55]. There is also strong evidence for synergy between EGF and TGFβ signaling pathways in cancer cell proliferation [55]. Smad7 is upregulated by both TGFβ and EGF pathways [56]. Hence, similar synergy may occur in the beta cell proliferation process, and that Smad7 is the key regulator in this synergy. Given that the TGFβ receptor signaling pathway prevents beta cell proliferation, EGF signaling may suppress proliferation prevention induced by TGFβ though AKT and MAPK signaling pathways.
5. Proposed Mechanism for TGFβ-Regulated Beta Cell Proliferation during Pregnancy
Fetal growth depends on nutrition from the mother through the placenta during pregnancy [57]. Glucose provides the necessary energy and is transported across the placenta via a passive process. In the early stages of fetal development, glucose transport depends on a beta cell-induced gradient between mother and fetus [58]. In the later stages of pregnancy, this glucose concentration gradient is threatened by the developing fetus, which diverts increasing amounts of glucose from the mother [58]. To maintain the proper glucose gradient, the placenta secretes hormones to increase maternal blood glucose via insulin resistance and increased hepatic glucose production [59,60]. In rodents, beta cell mass expansion in pregnancy results from beta cell proliferation from existing beta cells [61]. Interestingly, the pregnancy-associated increases in beta cell mass parallels the rise of placental lactogens [62,63]. Placental lactogens and prolactin treatment drive rodent beta cell proliferation efficiently via ERK1/2 activity, and this process depends on the activation of beta cell prolactin receptor (PRLR), which is induced by placental lactogens and prolactin. Nicole et al. found that TGFβ1 inhibits PRL expression via downstream Smad activation [64]. These pioneering studies suggest a possible role of TGFβ receptor signaling in control of parental beta cell growth.
6. Discussion
Whereas the intestine relies on specialized stem cells to replenish and repair tissue cells, a stem cell appears to be absent in the pancreas [65]. Instead, pancreas depends primarily on mature cell types for cell regeneration under both homeostatic and injury conditions. The key challenge for beta cell replacement and regeneration is inducing beta cell replication via manipulating cell cycle molecules and underlying regulatory pathways. Several approaches, such as overexpression of CDKs and cyclins or inhibition of CDKIs (p18INK4c, p21CIP1 and p27KIP1), may increase beta cell replication. TGFβ receptor signaling pathway is well known for its function as a regulator of normal development. Emerging research has revealed that the TGFβ receptor signaling pathway also plays a central role in beta cell proliferation and functions, including glucose stimulated insulin secretion, compensatory beta cell insulin gene transcription. While beta cell proliferation in T2D may directly contribute to increases in functional beta cell mass, newly born beta cells in T1D are exposed to the autoimmune environment to be attacked. Our recent publication showed that reprogrammed beta cells from alpha cells by gene therapy may form new beta cells that are slightly different from normal beta cells, which spare them from immune attack for a certain period [66]. Hence, forming immune-innocent beta cells is exceptionally important in T1D. A recent concept of rendering differentiated beta cells back to a less differentiated or a stem-like state, a process called dedifferentiation, was found to be necessary for beta cell proliferation [43,67,68,69,70,71,72,73]. Accumulation of our knowledge of the complex network governing beta cell proliferation may eventually lead to successful beta cell replacement therapy.
Funding
This research received no external funding.
Acknowledgments
X.X. was supported by a Tenure-track Assistant Professor startup from the Pediatric Division of Children’s Hospital of Pittsburgh.
Conflicts of Interest
The authors declare that they have no competing financial interests.
References
- Mallone, R.; Roep, B.O. Biomarkers for immune intervention trials in type 1 diabetes. Clin. Immunol. 2013, 149, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in european subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Positive and negative regulation of TGF-beta signaling. J. Cell Sci. 2000, 113, 1101–1109. [Google Scholar] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landstrom, M. TGF-beta promotes PI3K-AKT signaling and prostate cancer cell migration through the traf6-mediated ubiquitylation of p85alpha. Sci. Signal 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Gaffar, I.; Guo, P.; Wiersch, J.; Fischbach, S.; Peirish, L.; Song, Z.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; et al. M2 macrophages promote beta-cell proliferation by up-regulation of Smad7. Proc. Natl. Acad. Sci. USA 2014, 111, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Fischbach, S.; Song, Z.; Gaffar, I.; Zimmerman, R.; Wiersch, J.; Prasadan, K.; Shiota, C.; Guo, P.; Ramachandran, S.; et al. Transient suppression of TGFβ receptor signaling facilitates human islet transplantation. Endocrinology 2016, 157, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- El-Gohary, Y.; Tulachan, S.; Wiersch, J.; Guo, P.; Welsh, C.; Prasadan, K.; Paredes, J.; Shiota, C.; Xiao, X.; Wada, Y.; et al. A SMAD signaling network regulates islet cell proliferation. Diabetes 2014, 63, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. Beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.U.; Olewinski, M.; Tannapfel, A.; Schmidt, W.E.; Fritsch, H.; Meier, J.J. Cell cycle control of beta-cell replication in the prenatal and postnatal human pancreas. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Clemente, G.; Hu, J.; Pontecorvi, A.; Holst, J.J.; Giaccari, A.; Kulkarni, R.N. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014, 63, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.A.; Hollister-Lock, J.; Bonner-Weir, S.; Weir, G.C. Reduced Ki67 staining in the postmortem state calls into question past conclusions about the lack of turnover of adult human beta-cells. Diabetes 2015, 64, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Kushner, J.A. The role of aging upon beta cell turnover. J. Clin. Investig. 2013, 123, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. P16ink4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.G.; Dubus, P.; Mettus, R.V.; Galbreath, E.J.; Boden, G.; Reddy, E.P.; Barbacid, M. Loss of CDK4 expression causes insulin-deficient diabetes and CDK4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Cozar-Castellano, I.; Takane, K.K.; Bottino, R.; Balamurugan, A.N.; Stewart, A.F. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004, 53, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Salpeter, S.J.; Klochendler, A.; Weinberg-Corem, N.; Porat, S.; Granot, Z.; Shapiro, A.M.; Magnuson, M.A.; Eden, A.; Grimsby, J.; Glaser, B.; et al. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic beta-cells through glycolysis and calcium channels. Endocrinology 2011, 152, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi-Taesch, N.M.; Kleinberger, J.W.; Salim, F.G.; Troxell, R.; Wills, R.; Tanwir, M.; Casinelli, G.; Cox, A.E.; Takane, K.K.; Srinivas, H.; et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human beta-cell replication: A revised model of human beta-cell G1/S control. Diabetes 2013, 62, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi-Taesch, N.; Bigatel, T.A.; Sicari, B.; Takane, K.K.; Salim, F.; Velazquez-Garcia, S.; Harb, G.; Selk, K.; Cozar-Castellano, I.; Stewart, A.F. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for CDK-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes 2009, 58, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Mateo, C.M.; Marx, S.J. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J. Clin. Endocrinol. Metab. 2009, 94, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Hofler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi-Taesch, N.M.; Kleinberger, J.W.; Salim, F.G.; Troxell, R.; Wills, R.; Tanwir, M.; Casinelli, G.; Cox, A.E.; Takane, K.K.; Scott, D.K.; et al. Human pancreatic beta-cell g1/s molecule cell cycle atlas. Diabetes 2013, 62, 2450–2459. [Google Scholar] [CrossRef] [PubMed]
- Daneshmandi, S.; Karimi, M.H.; Pourfathollah, A.A. TGF-beta engineered mesenchymal stem cells (TGF-beta/MSCs) for treatment of type 1 diabetes (T1D) mice model. Int. Immunopharmacol. 2017, 44, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Grewal, K.D.; Wong, F.S.; Wang, H.; Picarella, D.E.; Janeway, C.A., Jr.; Flavell, R.A. Expression of transgene encoded TGF-beta in islets prevents autoimmune diabetes in nod mice by a local mechanism. J. Autoimmun. 2002, 19, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sanvito, F.; Nichols, A.; Herrera, P.L.; Huarte, J.; Wohlwend, A.; Vassalli, J.D.; Orci, L. TGF-beta 1 overexpression in murine pancreas induces chronic pancreatitis and, together with TNF-alpha, triggers insulin-dependent diabetes. Biochem. Biophys. Res. Commun. 1995, 217, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qi, S.; Hu, B.; Luo, H.; Wu, J. TGF-beta I promotes islet beta-cell function and regeneration. J. Immunol. 2011, 186, 5833–5844. [Google Scholar] [CrossRef] [PubMed]
- Bottinger, E.P.; Jakubczak, J.L.; Roberts, I.S.; Mumy, M.; Hemmati, P.; Bagnall, K.; Merlino, G.; Wakefield, L.M. Expression of a dominant-negative mutant TGF-beta type ii receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J. 1997, 16, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Crisera, C.A.; Rose, M.I.; Connelly, P.R.; Li, M.; Colen, K.L.; Longaker, M.T.; Gittes, G.K. The ontogeny of TGF-beta1, -beta2, -beta3, and TGF-beta receptor-ii expression in the pancreas: Implications for regulation of growth and differentiation. J. Pediatr. Surg. 1999, 34, 689–693; discussion 693–684. [Google Scholar] [CrossRef]
- Sanvito, F.; Herrera, P.L.; Huarte, J.; Nichols, A.; Montesano, R.; Orci, L.; Vassalli, J.D. TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development 1994, 120, 3451–3462. [Google Scholar] [PubMed]
- Xiao, X.; Wiersch, J.; El-Gohary, Y.; Guo, P.; Prasadan, K.; Paredes, J.; Welsh, C.; Shiota, C.; Gittes, G.K. TGFbeta receptor signaling is essential for inflammation-induced but not beta-cell workload-induced beta-cell proliferation. Diabetes 2013, 62, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Zhou, X.; Pang, Y.; Mao, Y.; Lu, X.; Li, M.; Zhang, J. TGF-beta signalling prevents pancreatic beta cell death after proliferation. Cell Prolif. 2015, 48, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Lingohr, M.K.; Dickson, L.M.; McCuaig, J.F.; Hugl, S.R.; Twardzik, D.R.; Rhodes, C.J. Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-alpha or EGF, augments pancreatic beta-cell proliferation. Diabetes 2002, 51, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Lee, J.H.; Yadav, H.; Kamaraju, A.K.; Liu, E.; Zhigang, D.; Vieira, A.; Kim, S.J.; Collins, H.; Matschinsky, F.; et al. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J. Biol. Chem. 2009, 284, 12246–12257. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, Y.; Tabuchi, M.; Kojima, I.; Eto, Y.; Shibai, H.; Ogata, E. Stimulation of insulin secretion by transforming growth factor-beta. Biochem. Biophys. Res. Commun. 1989, 158, 1060–1065. [Google Scholar] [CrossRef]
- Nomura, M.; Zhu, H.L.; Wang, L.; Morinaga, H.; Takayanagi, R.; Teramoto, N. Smad2 disruption in mouse pancreatic beta cells leads to islet hyperplasia and impaired insulin secretion due to the attenuation of ATP-sensitive k+ channel activity. Diabetologia 2014, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.G.; Apelqvist, A.A.; Gu, X.; Harmon, E.B.; Topper, J.N.; MacDonald, R.J.; Kim, S.K. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 2006, 4, e39. [Google Scholar] [CrossRef] [PubMed]
- Sjoholm, A.; Hellerstrom, C. TGF-beta stimulates insulin secretion and blocks mitogenic response of pancreatic beta-cells to glucose. Am. J. Physiol. 1991, 260, C1046–C1051. [Google Scholar] [CrossRef] [PubMed]
- Alman, A.C.; Talton, J.W.; Wadwa, R.P.; Urbina, E.M.; Dolan, L.M.; Hamman, R.F.; D′Agostino, R.B., Jr.; Marcovina, S.M.; Dabelea, D.M. Inflammation, adiposity, and progression of arterial stiffness in adolescents with type 1 diabetes: The search CVD study. J. Diabetes Complicat. 2018, 32, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Guadarrama-Lopez, A.L.; Valdes-Ramos, R.; Martinez-Carrillo, B.E. Type 2 diabetes, pufas, and vitamin d: Their relation to inflammation. J. Immunol. Res. 2014, 2014, 860703. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.V.; Shaw, L.C.; Grant, M.B. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012, 3, 170. [Google Scholar] [CrossRef] [PubMed]
- Sanjeevi, N.; Lipsky, L.M.; Nansel, T.R. Greater inflammation and adiposity are associated with lower bone mineral density in youth with type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 144, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Malik, A.B. NF-kappa b activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Yuan, M. Inflammation and the ikk beta/i kappa b/nf-kappa b axis in obesity- and diet-induced insulin resistance. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and specific NF-kappab blockade protects pancreatic beta cells from diabetogenic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D. Role of NF-kappab in beta-cell death. Biochem. Soc. Trans. 2008, 36, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Descargues, P.; Sil, A.K.; Sano, Y.; Korchynskyi, O.; Han, G.; Owens, P.; Wang, X.J.; Karin, M. Ikkalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Chen, F.; Kuang, C.; Chen, Y. Suppression of matrix metalloproteinase-9 transcription by transforming growth factor-beta is mediated by a nuclear factor-kappab site. Biochem. J. 2004, 381, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Alejandro, E.U. Control of pancreatic beta-cell fate by insulin signaling: The sweet spot hypothesis. Cell Cycle 2008, 7, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H., 3rd; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. Foxo1 protects against pancreatic beta cell failure through neurod and mafa induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Zarrouki, B.; Benterki, I.; Fontes, G.; Peyot, M.L.; Seda, O.; Prentki, M.; Poitout, V. Epidermal growth factor receptor signaling promotes pancreatic beta-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes 2014, 63, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocana, A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Uttamsingh, S.; Bao, X.; Nguyen, K.T.; Bhanot, M.; Gong, J.; Chan, J.L.; Liu, F.; Chu, T.T.; Wang, L.H. Synergistic effect between EGF and TGF-beta1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene 2008, 27, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Afrakhte, M.; Moren, A.; Jossan, S.; Itoh, S.; Sampath, K.; Westermark, B.; Heldin, C.H.; Heldin, N.E.; ten Dijke, P. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem. Biophys. Res. Commun. 1998, 249, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W., Jr. Energy and substrate requirements of the placenta and fetus. Proc. Nutr. Soc. 1991, 50, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.U.; Deborde, S.; Illsley, N.P. Placental glucose transfer and fetal growth. Endocrine 2002, 19, 13–22. [Google Scholar] [CrossRef]
- Espinosa de los, M.; Driscoll, S.G.; Steinke, J. Insulin release from isolated human fetal pancreatic islets. Science 1970, 168, 1111–1112. [Google Scholar] [CrossRef]
- Lain, K.Y.; Catalano, P.M. Metabolic changes in pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Bone, A.J.; Taylor, K.W. Mitabolic adaptation to pregnancy shown by increased biosynthesis of insulin in islets of langerhans isolated from pregnant rat. Nature 1976, 262, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.A.; Brelje, T.C.; Sorenson, R.L. Adaptation of islets of langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130, 1459–1466. [Google Scholar] [PubMed]
- Van Assche, F.A.; Aerts, L.; De Prins, F. A morphological study of the endocrine pancreas in human pregnancy. Br. J. Obstet. Gynaecol. 1978, 85, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Kane, N.M.; Jones, M.; Brosens, J.J.; Kelly, R.W.; Saunders, P.T.; Critchley, H.O. TGFβ1 attenuates expression of prolactin and IGFbp-1 in decidualized endometrial stromal cells by both Smad-dependent and Smad-independent pathways. PLoS ONE 2010, 5, e12970. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous reprogramming of alpha cells into beta cells, induced by viral gene therapy, reverses autoimmune diabetes. Cell Stem Cell 2018, 22, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.H.; Sagar; Arrigoni, L.; Dalgaard, K.; et al. The polycomb-dependent epigenome controls beta cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018, 27, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Knoch, K.P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.C.; Bornstein, S.R.; et al. Virus-like infection induces human beta cell dedifferentiation. JCI Insight 2018, 3, 97732. [Google Scholar] [CrossRef] [PubMed]
- Efrat, S. Mechanisms of adult human beta-cell in vitro dedifferentiation and redifferentiation. Diabetes Obes. Metab. 2016, 18, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Fiori, J.L.; Shin, Y.K.; Kim, W.; Krzysik-Walker, S.M.; Gonzalez-Mariscal, I.; Carlson, O.D.; Sanghvi, M.; Moaddel, R.; Farhang, K.; Gadkaree, S.K.; et al. Resveratrol prevents beta-cell dedifferentiation in nonhuman primates given a high-fat/high-sugar diet. Diabetes 2013, 62, 3500–3513. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Landsman, L.; Parent, A.; Hebrok, M. Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 17010–17015. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Canonical TGFβ/SMAD signaling pathway. TGFβ binds to type II receptors on the cell membrane, recruiting type I receptors to phosphorylate Smad2 and Smad3 (R-Smad) to form a complex with Smad4. The complex then translocates from the cytoplasm to the nucleus and regulate downstream gene expression.
Figure 1.
Canonical TGFβ/SMAD signaling pathway. TGFβ binds to type II receptors on the cell membrane, recruiting type I receptors to phosphorylate Smad2 and Smad3 (R-Smad) to form a complex with Smad4. The complex then translocates from the cytoplasm to the nucleus and regulate downstream gene expression.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jiang, Y.; Fischbach, S.; Xiao, X. The Role of the TGFβ Receptor Signaling Pathway in Adult Beta Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3136. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103136
AMA Style
Jiang Y, Fischbach S, Xiao X. The Role of the TGFβ Receptor Signaling Pathway in Adult Beta Cell Proliferation. International Journal of Molecular Sciences. 2018; 19(10):3136. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103136
Chicago/Turabian StyleJiang, Yinan, Shane Fischbach, and Xiangwei Xiao. 2018. "The Role of the TGFβ Receptor Signaling Pathway in Adult Beta Cell Proliferation" International Journal of Molecular Sciences 19, no. 10: 3136. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103136
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.