The Interaction of Selenium with Chemotherapy and Radiation on Normal and Malignant Human Mononuclear Blood Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
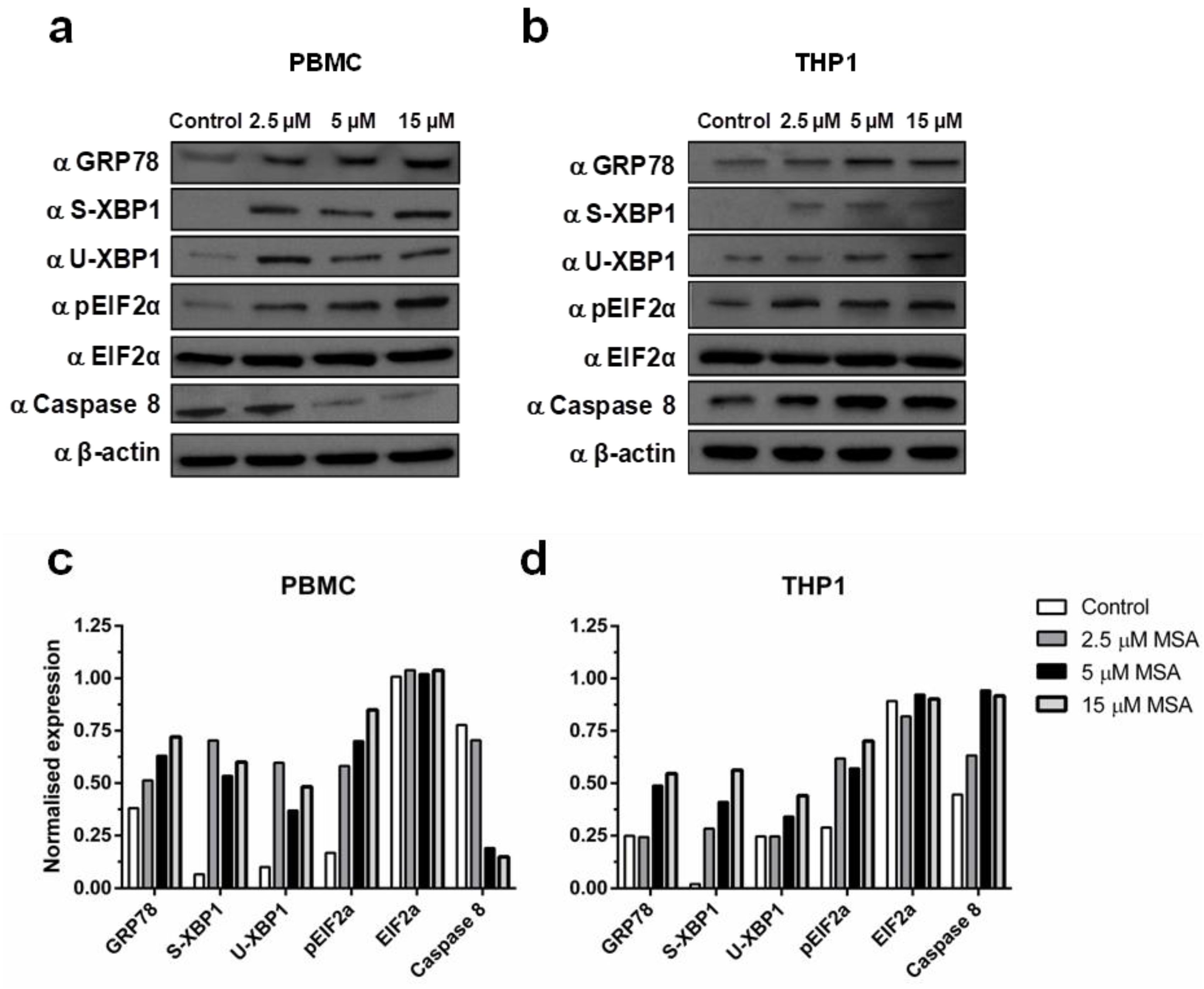
2.1. Methylseleninic Acid (MSA) Induces Endoplasmic Reticulum (ER) Stress in Normal and Malignant Cells But Differentially Modulates Apoptosis
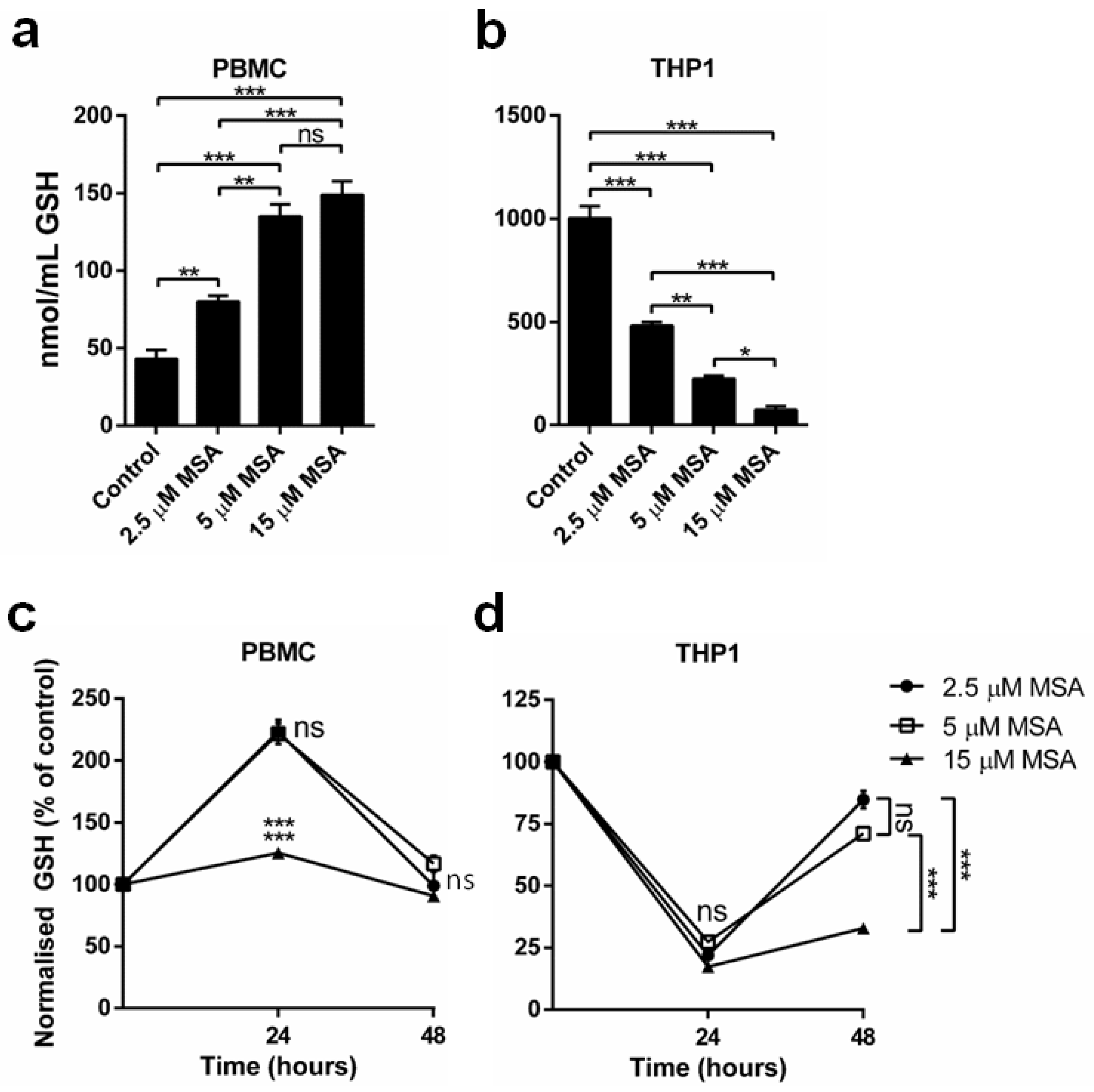
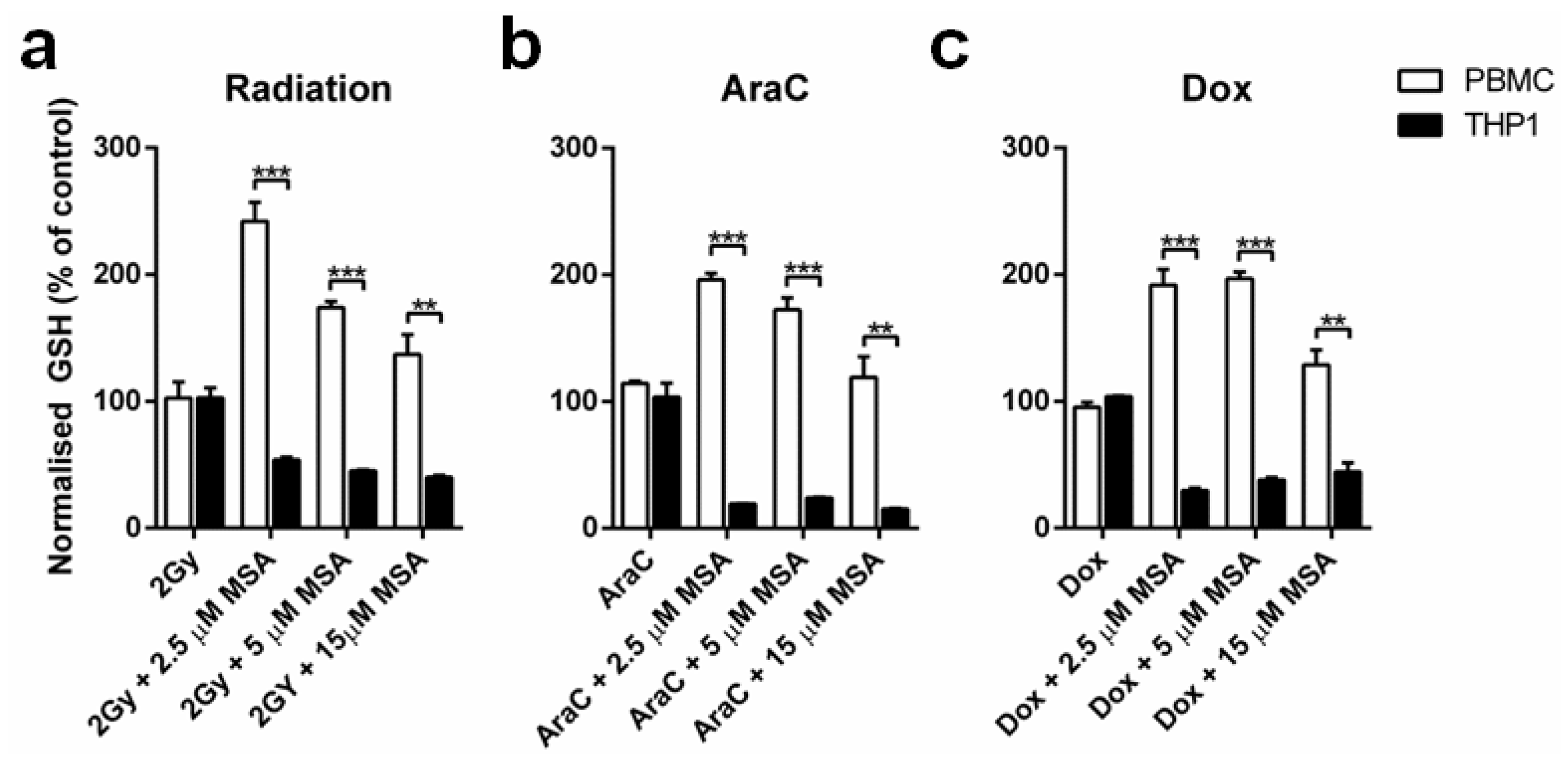
2.2. MSA Has a Divergent Impact on Glutathione (GSH) Levels in Normal and Malignant Cells
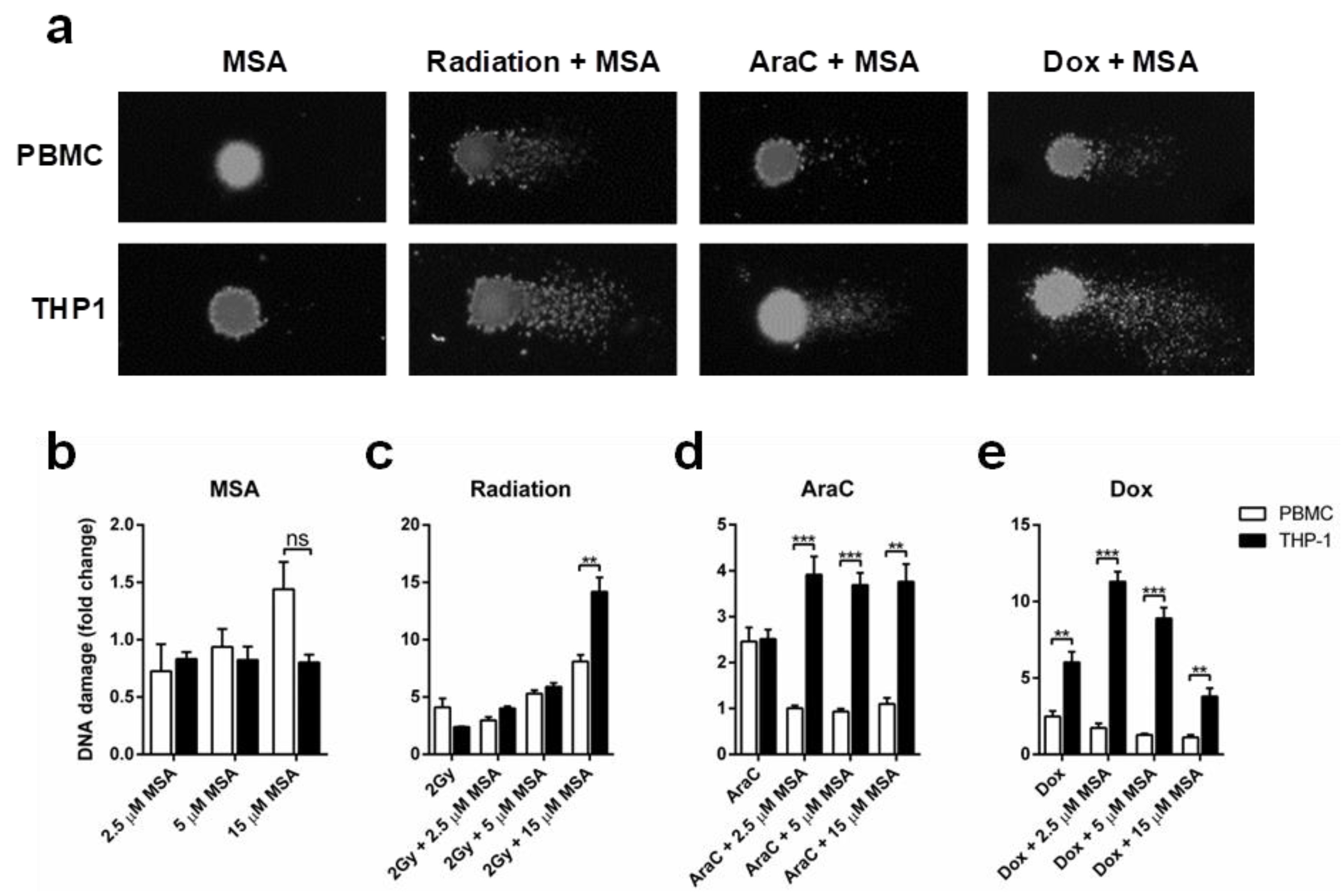
2.3. MSA Reduces DNA Damage in Normal Cells While Increasing DNA Damage in Malignant Cells
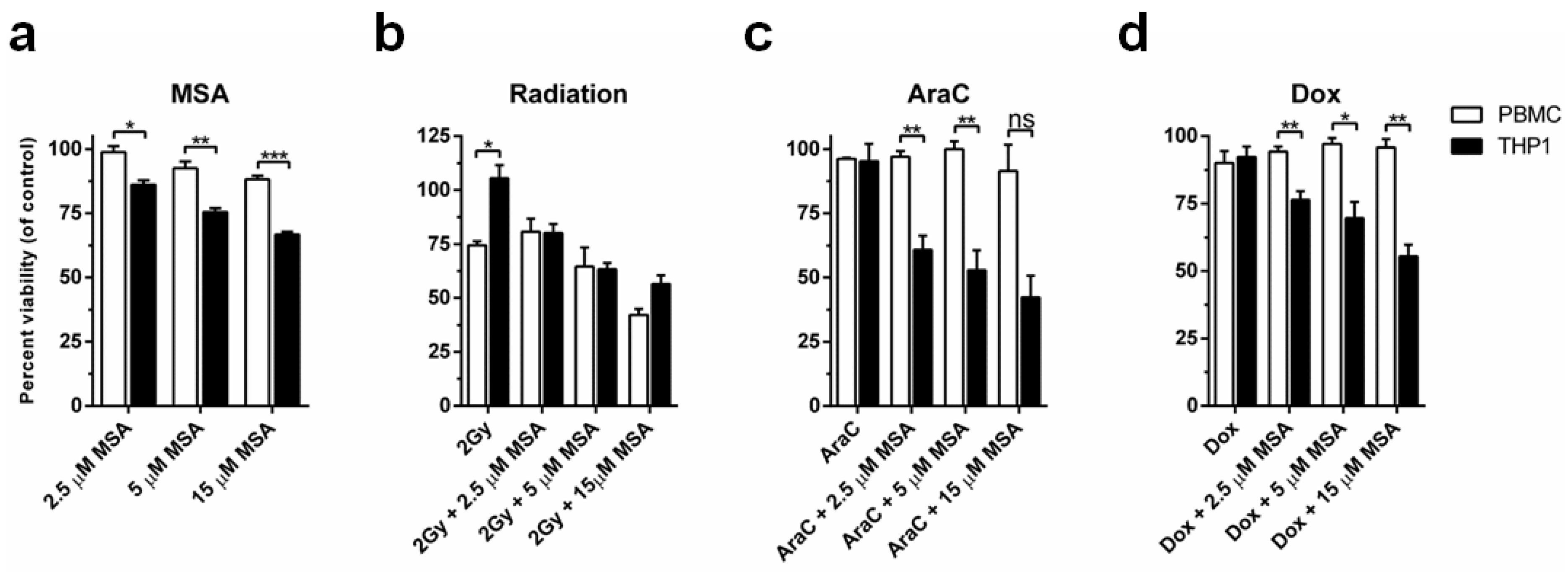
2.4. MSA Treatment Protects Normal Cells While Potentiating Cell Death in Malignant Cells
3. Discussion
4. Materials and Methods
4.1. Mononuclear Cell Isolation
4.2. Cell Culture
4.3. Western Blot Analysis
4.4. Measurement of GSH
4.5. Comet Assay
4.6. MTT Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AraC | Cytosine arabinoside |
| Dox | Doxorubicin |
| GSH | Glutathione |
| MSA | Methylseleninic acid |
| PBMC | Peripheral blood mononuclear cell |
References
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Lipinski, B. Sodium Selenite as an Anticancer Agent. Anticancer Agents Med. Chem. 2017, 17, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.O.; Khairuddin, P.F.; Jameson, M.B. Optimising Selenium for Modulation of Cancer Treatments. Anticancer Res. 2017, 37, 6497–6509. [Google Scholar] [PubMed]
- Cao, S.; Durrani, F.A.; Rustum, Y.M. Selective Modulation of the Therapeutic Efficacy of Anticancer Drugs by Selenium Containing Compounds against Human Tumor Xenografts. Clin. Cancer Res. 2004, 10, 2561–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.; Cao, S.; Durrani, F.A.; Rustum, Y.M. Selenium protects against toxicity induced by anticancer drugs and augments antitumor activity: A highly selective, new, and novel approach for the treatment of solid tumors. Clin. Colorectal Cancer 2005, 5, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Francescato, H.D.; Costa, R.S.; Camargo, S.M.R.; Zanetti, M.A.; Lavrador, M.A.; Bianchi, M.D. Effect of oral selenium administration on cisplatin-induced nephrotoxicity in rats. Pharmacol. Res. 2001, 43, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Kumaran, M.N.; Gounder, M.; Gibbon, D.G.; Nieves-Neira, W.; Vaidya, A.; Hellmann, M.; Kane, M.P.; Buckley, B.; Shih, W.; et al. Phase I trial of selenium plus chemotherapy in gynecologic cancers. Gynecol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Puspitasari, I.M.; Abdulah, R.; Yamazaki, C.; Kameo, S.; Nakano, T.; Koyama, H. Updates on clinical studies of selenium supplementation in radiotherapy. Radiat. Oncol. 2014, 9, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieja, K.; Talerczyk, M. Selenium as an element in the treatment of ovarian cancer in women receiving chemotherapy. Gynecol. Oncol. 2004, 93, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Asfour, I.A.; El Tehewi, M.M.; Ahmed, M.H.; Abdel-Sattar, M.A.; Moustafa, N.N.; Hegab, H.M.; Fathey, O.M. High-dose sodium selenite can induce apoptosis of lymphoma cells in adult patients with non-Hodgkin’s lymphoma. Biol. Trace Elem. Res. 2009, 127, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Asfour, I.A.; Fayek, M.; Raouf, S.; Soliman, M.; Hegab, H.M.; El Desoky, H.; Saleh, R.; Moussa, M.A. The impact of high-dose sodium selenite therapy on Bcl-2 expression in adult non-Hodgkin’s lymphoma patients: Correlation with response and survival. Biol. Trace Elem. Res. 2007, 120, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Asfour, I.A.; El Shazly, S.; Fayek, M.H.; Hegab, H.M.; Raouf, S.; Moussa, M.A. Effect of high-dose sodium selenite therapy on polymorphonuclear leukocyte apoptosis in non-Hodgkin’s lymphoma patients. Biol. Trace Elem. Res. 2006, 110, 19–32. [Google Scholar] [CrossRef]
- Hu, Y.J.; Chen, Y.; Zhang, Y.Q.; Zhou, M.Z.; Song, X.M.; Zhang, B.Z.; Luo, L.; Xu, P.M.; Zhao, Y.N.; Zhao, Y.B.; et al. The protective role of selenium on the toxicity of cisplatin-contained chemotherapy regimen in cancer patients. Biol. Trace Elem. Res. 1997, 56, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Jahangard-Rafsanjani, Z.; Gholami, K.; Hadjibabaie, M.; Shamshiri, A.R.; Alimoghadam, K.; Sarayani, A.; Mojtahedzadeh, M.; Ostadali-Dehaghi, M.; Ghavamzadeh, A. The efficacy of selenium in prevention of oral mucositis in patients undergoing hematopoietic SCT: A randomized clinical trial. Bone Marrow Transplant. 2013, 48, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Muecke, R.; Schomburg, L.; Glatzel, M.; Berndt-Skorka, R.; Baaske, D.; Reichl, B.; Buentzel, J.; Kundt, G.; Prott, F.J.; Devries, A.; et al. Multicenter, phase 3 trial comparing selenium supplementation with observation in gynecologic radiation oncology. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Buntzel, J.; Riesenbeck, D.; Glatzel, M.; Berndt-Skorka, R.; Riedel, T.; Mucke, R.; Kisters, K.; Schonekaes, K.G.; Schafer, U.; Bruns, F.; et al. Limited effects of selenium substitution in the prevention of radiation-associated toxicities. Results of a randomized study in head and neck cancer patients. Anticancer Res. 2010, 30, 1829–1832. [Google Scholar] [PubMed]
- Muecke, R.; Micke, O.; Schomburg, L.; Buentzel, J.; Kisters, K.; Adamietz, I.A. Selenium in Radiation Oncology-15 Years of Experiences in Germany. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.C.; Doll, D.C.; Freter, C.E. Perry’s The Chemotherapy Souce Book, 5th ed.; Lippincott, Williams & Wilkins: New York, NY, USA, 2012. [Google Scholar]
- Mishra, K.; Alsbeih, G. Appraisal of biochemical classes of radioprotectors: Evidence, current status and guidelines for future development. 3Biotech 2017, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Devine, A.; Marignol, L. Potential of Amifostine for Chemoradiotherapy and Radiotherapy-associated Toxicity Reduction in Advanced NSCLC: A Meta-Analysis. Anticancer Res. 2016, 36, 5–12. [Google Scholar] [PubMed]
- Freyer, D.R.; Chen, L.; Krailo, M.D.; Knight, K.; Villaluna, D.; Bliss, B.; Pollock, B.H.; Ramdas, J.; Lange, B.; Van, H.D.; et al. Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 63–74. [Google Scholar] [CrossRef]
- Liu, M.; Hu, C.; Xu, Q.; Chen, L.; Ma, K.; Xu, N.; Zhu, H. Methylseleninic acid activates Keap1/Nrf2 pathway via up-regulating miR-200a in human oesophageal squamous cell carcinoma cells. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Bukur, J.; Hochgrafe, F.; Wessjohann, L.A.; Lichtenfels, R.; Seliger, B. Modulation of MHC class I surface expression in B16F10 melanoma cells by methylseleninic acid. Oncoimmunology 2017, 6, e1259049. [Google Scholar] [CrossRef] [PubMed]
- Kassam, S.; Goenaga-Infante, H.; Maharaj, L.; Hiley, C.T.; Juliger, S.; Joel, S.P. Methylseleninic acid inhibits HDAC activity in diffuse large B-cell lymphoma cell lines. Cancer Chemother. Pharmacol. 2011, 68, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Dong, Y.; Ganther, H.E. New concepts in selenium chemoprevention. Cancer Metastasis Rev. 2002, 21, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [PubMed]
- Zakharia, Y.; Bhattacharya, A.; Rustum, Y.M. Selenium targets resistance biomarkers enhancing efficacy while reducing toxicity of anti-cancer drugs: Preclinical and clinical development. Oncotarget 2018, 9, 10765–10783. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, H.; Dong, Y.; Park, Y.M.; Ip, C. Endoplasmic reticulum stress signal mediators are targets of selenium action. Cancer Res. 2005, 65, 9073–9079. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, H.; Li, Y.; Wu, Z.; Zhu, Y.; Wang, T.; Gao, A.C.; Chen, J.; Zhou, Q. Intracellular glutathione content influences the sensitivity of lung cancer cell lines to methylseleninic acid. Mol. Carcinog. 2012, 51, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Ding, W.X.; Ong, C.N. Intracellular glutathione is a cofactor in methylseleninic acid-induced apoptotic cell death of human hepatoma HEPG(2) cells. Free Radic. Biol. Med. 2002, 33, 552–561. [Google Scholar] [CrossRef]
- Caffrey, P.B.; Frenkel, G.D. Selenium compounds prevent the induction of drug resistance by cisplatin in human ovarian tumor xenografts in vivo. Cancer Chemother. Pharmacol. 2000, 46, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.L.; Mihelc, E.M.; Pollok, K.E.; Smith, M.L. Chemotherapeutic selectivity conferred by selenium: A role for p53-dependent DNA repair. Mol. Cancer Ther. 2007, 6, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.R.; Sweeney, C.; Smith, M.L. Selenomethionine induction of DNA repair response in human fibroblasts. Oncogene 2002, 21, 3663–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.H.; Yoon, M.J.; Kim, M.; Kim, J.I.; Lee, S.J.; Lee, Y.S.; Bae, S. Enhanced lung cancer cell killing by the combination of selenium and ionizing radiation. Oncol. Rep. 2007, 17, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Carrier, L.; Rowan, B.G. Methylseleninic acid synergizes with tamoxifen to induce caspase-mediated apoptosis in breast cancer cells. Mol. Cancer Ther. 2008, 7, 3056–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Wang, Z.; Ganther, H.; Lu, J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001, 61, 3062–3070. [Google Scholar] [PubMed]
- Li, S.; Zhou, Y.; Wang, R.; Zhang, H.; Dong, Y.; Ip, C. Selenium sensitizes MCF-7 breast cancer cells to doxorubicin-induced apoptosis through modulation of phospho-Akt and its downstream substrates. Mol. Cancer Ther. 2007, 6, 1031–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.H.; Evens, A.M. Oxidative stress and apoptosis: A new treatment paradigm in cancer. Front. Biosci. 2006, 11, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.W.; Meyn, R.E. Redox aspects of Bcl-2 function. Antioxid. Redox Signal. 2000, 2, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Biaglow, J.E.; Varnes, M.E.; Epp, E.R.; Clark, E.P.; Tuttle, S.W.; Held, K.D. Role of glutathione and other thiols in cellular response to radiation and drugs. Drug Metab. Rev. 1989, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bump, E.A.; Brown, J.M. Role of glutathione in the radiation response of mammalian cells in vitro and in vivo. Pharmacol. Ther. 1990, 47, 117–136. [Google Scholar] [CrossRef]
- Coleman, C.N.; Bump, E.A.; Kramer, R.A. Chemical modifiers of cancer treatment. J. Clin. Oncol. 1988, 6, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Russo, A. The role of glutathione in radiation and drug induced cytotoxicity. Br. J. Cancer Suppl. 1987, 8, 96–104. [Google Scholar] [PubMed]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Vadgama, J.V.; Wu, Y.; Shen, D.; Hsia, S.; Block, J. Effect of selenium in combination with Adriamycin or Taxol on several different cancer cells. Anticancer Res. 2000, 20, 1391–1414. [Google Scholar] [PubMed]
- Valdiglesias, V.; Pasaro, E.; Mendez, J.; Laffon, B. In vitro evaluation of selenium genotoxic, cytotoxic, and protective effects: A review. Arch. Toxicol. 2010, 84, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and Toxicity of Sodium Selenite in the Treatment of Patients with Carcinoma in a Phase I Clinical Trial: The SECAR Study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, N.M.; Hovens, C.M.; Michael, M.; Rosenthal, M.A.; Costello, A.J. Open-label, phase I dose-escalation study of sodium selenate, a novel activator of PP2A, in patients with castration-resistant prostate cancer. Br. J. Cancer 2010, 103, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobb, R.J.; van, A.R.; Wiegmans, A.; Ham, S.; Larsen, J.E.; Moller, A. Exosomes derived from mesenchymal non-small cell lung cancer cells promote chemoresistance. Int. J. Cancer 2017, 141, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Evans, S.O.; Jacobson, G.M.; Goodman, H.J.B.; Bird, S.; Jameson, M.B. Comparative safety and pharmacokinetic evaluation of three oral selenium compounds in cancer patients. Biol. Trace Elem. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.R.; Burk, R.F.; Payne, O.R.; Hill, K.E.; Perloff, M.; Davis, W.; Pili, R.; George, S.; Bergan, R. Selenomethionine and methyl selenocysteine: Multiple-dose pharmacokinetics in selenium-replete men. Oncotarget 2017, 8, 26312–26322. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobb, R.J.; Jacobson, G.M.; Cursons, R.T.; Jameson, M.B. The Interaction of Selenium with Chemotherapy and Radiation on Normal and Malignant Human Mononuclear Blood Cells. Int. J. Mol. Sci. 2018, 19, 3167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103167
Lobb RJ, Jacobson GM, Cursons RT, Jameson MB. The Interaction of Selenium with Chemotherapy and Radiation on Normal and Malignant Human Mononuclear Blood Cells. International Journal of Molecular Sciences. 2018; 19(10):3167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103167
Chicago/Turabian StyleLobb, Richard J., Gregory M. Jacobson, Ray T. Cursons, and Michael B. Jameson. 2018. "The Interaction of Selenium with Chemotherapy and Radiation on Normal and Malignant Human Mononuclear Blood Cells" International Journal of Molecular Sciences 19, no. 10: 3167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103167