PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation


1
Department of Pediatrics, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 23142, Taiwan
2
Institute of Biomedical Sciences, Academia Sinica, Taipei 11529, Taiwan
3
Department of Pediatrics, Tzu Chi University, Hualien 97004, Taiwan
4
Institute of Pharmacology, Taipei Medical University, 250 Wu-Hsing Street, Taipei 11031, Taiwan
5
Department of Physiology, School of Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(11), 3447; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113447
Submission received: 31 August 2018
/
Revised: 23 October 2018
/
Accepted: 30 October 2018
/
Published: 2 November 2018
(This article belongs to the Special Issue PPARs in Cellular and Whole Body Energy Metabolism)
Abstract
:Traditionally, peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a 91 kDa transcription factor, regulates lipid metabolism and long-chain fatty acid oxidation by upregulating the expression of several genes of the tricarboxylic acid cycle and the mitochondrial fatty acid oxidation pathway. In addition, PGC-1α regulates the expression of mitochondrial genes to control mitochondria DNA replication and cellular oxidative metabolism. Recently, new insights showed that several myokines such as irisin and myostatin are epigenetically regulated by PGC-1α in skeletal muscles, thereby modulating systemic energy balance, with marked expansion of mitochondrial volume density and oxidative capacity in healthy or diseased myocardia. In addition, in our studies evaluating whether PGC-1α overexpression in epicardial adipose tissue can act as a paracrine organ to improve or repair cardiac function, we found that overexpression of hepatic PGC-1α increased hepatic fatty acid oxidation and decreased triacylglycerol storage and secretion in vivo and in vitro. In this review, we discuss recent studies showing that PGC-1α may regulate mitochondrial fusion–fission homeostasis and affect the renal function in acute or chronic kidney injury. Furthermore, PGC-1α is an emerging protein with a biphasic role in cancer, acting both as a tumor suppressor and a tumor promoter and thus representing a new and unresolved topic for cancer biology studies. In summary, this review paper demonstrates that PGC-1α plays a central role in coordinating the gene expression of key components of mitochondrial biogenesis and as a critical metabolic regulator in many vital organs, including white and brown adipose tissue, skeletal muscle, heart, liver, and kidney.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family includes ligands of multiple nuclear or non-nuclear receptors that control the expression of specific genes regulating cell metabolism. The first discovered member of the PGC-1 family, a 91 kDa nuclear protein [1] identified in brown adipose tissue (BAT) in mouse studies of cold-induced thermogenesis, was called peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α (PGC-1α) [2]. The biological activity of PGC-1α is tightly controlled at several levels: by transcriptional control (of multiple promoter regions), alternative splicing of transcripts, and post-translational modification (e.g., phosphorylation, acetylation, or methylation). This activity results in several mRNA isoforms—PGC-1α-a, PGC-1α-b, PGC-1α-c, and NTPGC-1α—that enable cellular adaptation to various environmental conditions [3]. Studies have shown that PGC-1α can be used in different tissues with different coactivators to induce changes in lipid oxidation, energy homeostasis, mitochondrial mass, and insulin sensitivity. Here, we review these studies.
2. PGC-1α Can Regulate Lipid Metabolism
As a transcription factor, PGC-1α can bind to targets such as PPARα, PPARβ/δ, and PPARγ, which coordinate the expression of mitochondrial genes and indirectly contribute to fatty acid (FA) transport and utilization [4]. Furthermore, PGC-1α upregulates the expression of several genes of the tricarboxylic acid cycle [5] and the mitochondrial FA oxidation pathway [6]. PGC-1α also regulates the expression of nuclear and mitochondrial genes that encode components of the electron transport system and oxidative phosphorylation (OXPHOS) via nuclear respiratory factors 1 and 2 (NRF-1 and -2) and estrogen-related receptor α (ERRα) coactivation. These effects can increase the expression of mitochondrial transcription factor A (mtTFA), which is known to control mtDNA replication and transcription and therefore regulate cellular oxidative metabolism [7]. Accordingly, the augmented expression of cytochrome c, cytochrome-c-oxidase subunits II and IV, and adenosine triphosphate (ATP) synthase also result from PGC-1α activation [8,9,10,11].
Another noteworthy effect of PGC-1α is its ability to stimulate peroxisomal activity such as the oxidation of long-chain and very-long-chain FAs [12]. Briefly, PGC-1α level is positively correlated with the ability of cells to fully oxidize FA, an effect that may reduce intramuscular lipid deposition and improve tissue insulin sensitivity. Chromatin immunoprecipitation assays have shown that the mechanism of this effect includes the coactivation of liver X receptor α (LXRα), which stimulates PGC-1α binding to the LXR response element in the FAS promoter. In addition, muscle-specific PGC-1α expression in MPGC-1α transgenic mice exacerbated de novo free fatty acid (FFA) synthesis as well as FA esterification and triacylglycerol (TAG) accumulation [13]. Furthermore, PGC-1α is involved in lipid distribution and may upregulate FAT/CD36, FABPpm, and FATP1 mRNA and protein expression in mitochondrial fractions. The latter effect was confirmed solely in murine FAT/CD36 and FABP3 cells [14].
3. PGC-1α as a Coactivator for Metabolic Homeostasis in Skeletal Muscle
Muscle adjusts to endurance exercise by promoting mitochondrial biogenesis, angiogenesis, and changes of fiber composition [15,16,17]. Chinsomboona et al., had reported that mice lacking PGC-1α in skeletal muscle failed to increase capillary density in response to exercise. This study showed that β-adrenergic stimulation of a PGC-1 α/estrogen-related receptor alpha (ERRα)/vascular endothelial growth factor (VEGF) axis modulates exercise-induced angiogenesis in skeletal muscle [18] and truncated PGC-1α can lead to hypoxic induction of VEGF and angiogenesis in skeletal muscle [19]. In addition, PGC-1α activates transcription in cooperation with myocyte enhancer factor-2 (Mef2) and acts as a target for calcineurin signaling, which has been involved in slow fiber gene expression [20]. Skeletal muscle-specific PGC-1α knock-out mice demonstrate a shift from oxidative type I and IIa toward type IIx and IIb glycolytic muscle fibers [21]. Rasbach et al., reported that PGC-1α–mediated switch to slow, oxidative fibers in vitro is dependent on hypoxia-inducible factor 2 α (HIF2α), and mice lacking HIF2α in muscle increase the expression of genes and proteins related to a fast-twitch-fiber-type switch [22]. Transgenic mice with mildly elevated muscle levels of PGC1α are also resistant to age-related obesity and diabetes and show a prolonged lifespan [23]. These results strongly suggest that PGC1α expression in skeletal muscles can significantly contribute to regulating systemic energy balance. Recent studies also demonstrated that muscle contraction may induce the secretion of molecules called myokines, which enables the crosstalk between skeletal muscle and other organs such as adipose tissue, bone, liver, kidney, and brain; in this sense, skeletal muscle can be considered an endocrine organ. Indeed, several myokines discovered in the past decade via secretome analysis include interleukin-6, irisin/fibronectin type III domain-containing protein 5 (FNDC5), myostatin, interleukin-15, brain-derived neurotrophic factor (BDNF), β-aminoisobutyric acid, meteorin-like, leukemia inhibitory factor, and secreted protein acidic and rich in cysteine (SPARC).
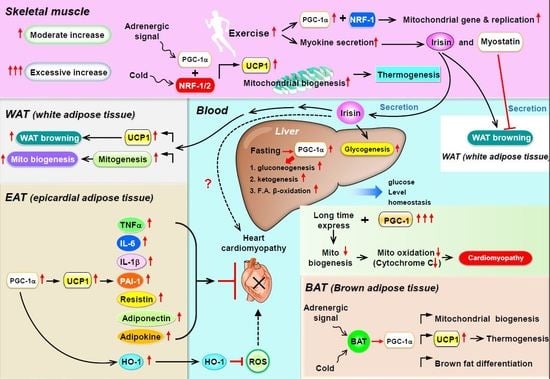
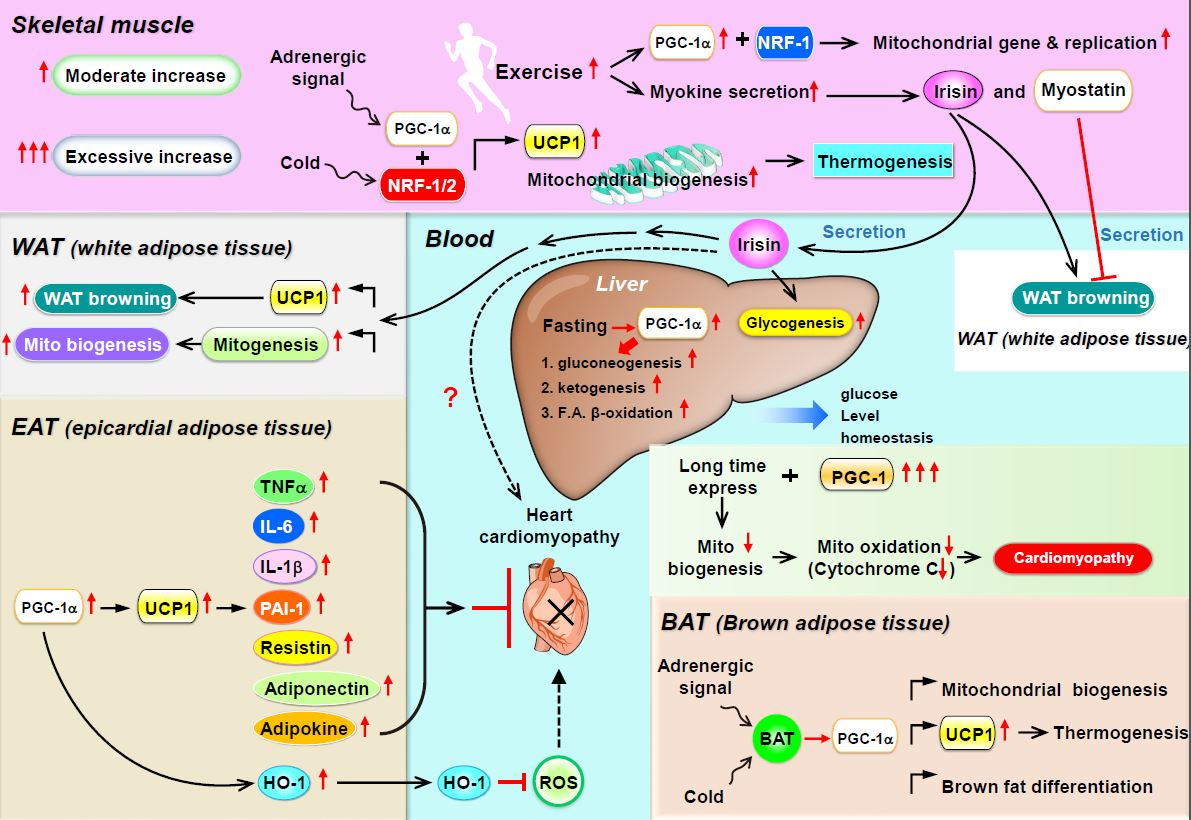
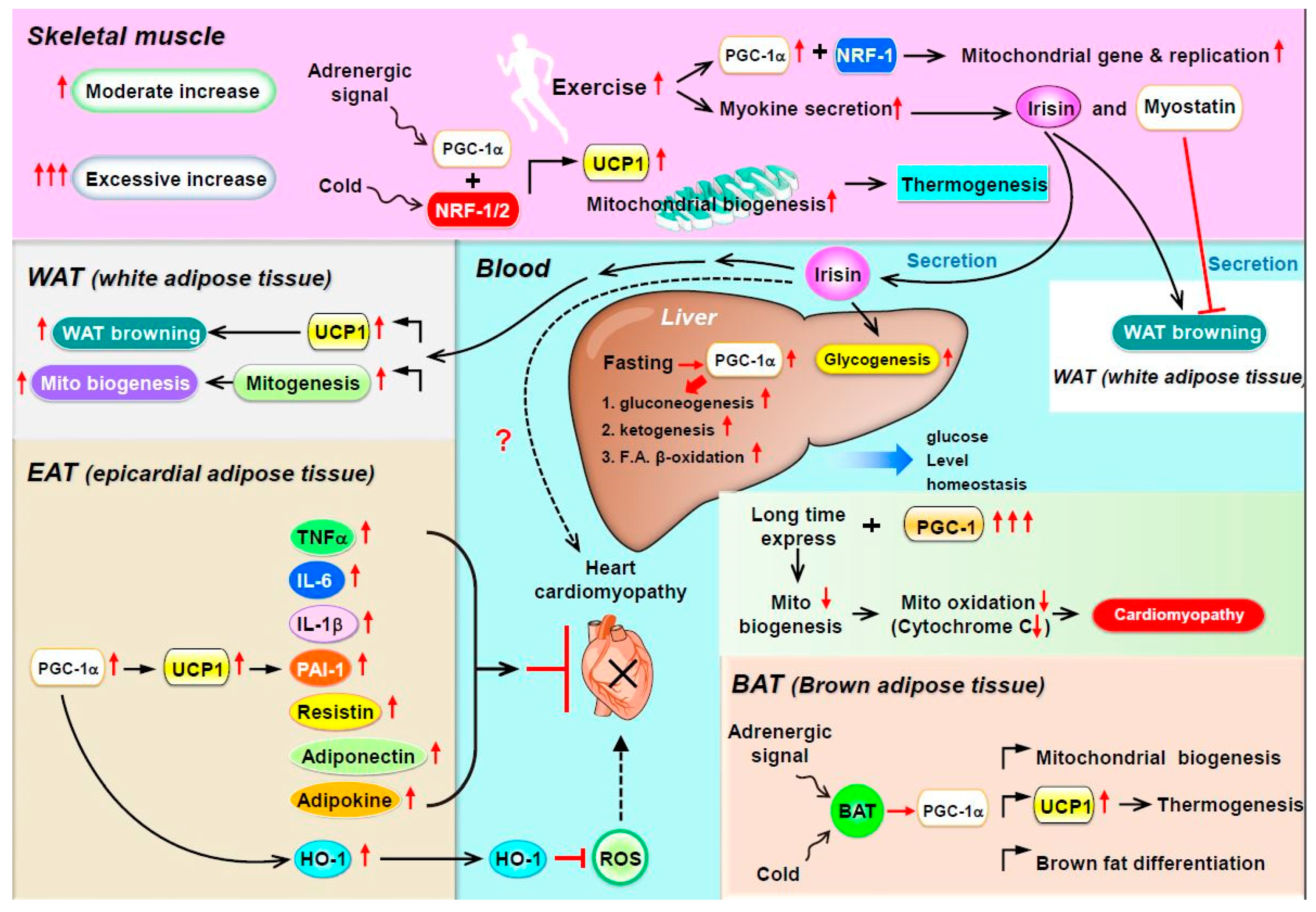
Several myokines are regulated by PGC-1: irisin/FNDC5, myostatin, and BDNF [24]. (1) Irisin is a PGC-1α-dependent myokine. In mice with muscle-specific PGC-1α overexpression, PGC-1α induces the expression of a membrane protein, FNDC5, and exercise triggers the cleavage of FNDC5 to generate irisin and then secreted into the bloodstream, which elevates energy expenditure in subcutaneous adipose tissue via adipocyte browning [25]. This process implies that PGC-1α overexpression with exercise may increase the expression of uncoupling protein 1 (UCP-1) and eventually increase the browning of white fat cells [25]. Recently, mass spectrometry was used to measure circulating irisin levels in humans in an antibody-independent manner; irisin levels were increased by both short and prolonged period exercise [26,27]. Under physiological conditions, irisin stimulates glucose uptake and lipid metabolism via the activation of AMP-activated protein kinase (AMPK) [28,29,30] and is also involved in muscle growth by inducing insulin-like growth factor 1 and suppressing myostatin [31]. In addition to having effects on muscle, exogenous administration of irisin induces adipocyte browning in subcutaneous fat in mice via p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase 1/2 (ERK1/2) [32]. In the murine liver, irisin stimulates glycogenesis but reduces gluconeogenesis and lipogenesis by regulating GSK3, FOXO1, and SREBP2 [33,34,35]. (2) Myostatin is an autocrine and paracrine hormone secreted by muscle fibers and the only myokine with inhibited secretion during muscle contraction and exercise [36]. In addition to its local involvement in muscle atrophy [37], myostatin can also modulate metabolic homeostasis by regulating adipose tissue function [38,39,40]. The inhibition of myostatin was found to ameliorate the development of obesity and insulin resistance in mice fed a high-fat diet, presumably by mechanisms promoting lipolysis and mitochondrial lipid oxidation in adipose tissue and liver [41]. In addition, Dong et al., showed that inhibition of myostatin resulted in the conversion of white adipose tissue (WAT) to brown adipose tissue (BAT), while enhancing fatty acid oxidation and increasing energy expenditure. Inhibition of myostatin increased PGC-1α expression and irisin production in muscle. Irisin stimulated browning via mediating muscle-to-fat cross talk [42]. Myostatin knockout mice are characterized by increased expression and phosphorylation of AMPK in muscle, which subsequently activates PGC1α and Fndc5. This study demonstrated that Fndc5 is upregulated and secreted from muscle to induce browning of WAT in myostatin knockout mice [43]. (3) BDNF is known primarily as a molecule released by the hypothalamus and as a key element regulating neuronal development, plasticity, and energy homeostasis [44]. Cao et al., found that hypothalamic overexpression of BDNF via recombinant adeno-associated virus (rAAV) duplicated the enriched environment (EE)-associated activation of the brown fat program and lean phenotype. This study suggested that induction of hypothalamic BDNF expression in response to environmental stimuli results in selective sympathoneural regulation of white fat browning and increased energy dissipation [45]. Wrann et al., showed hippocampal BDNF gene expression [46]. PGC-1α knockout mice show decreased FNDC5 expression in the brain. Overexpression of FNDC5 increases BDNF expression in primary cortical neurons. Furthermore, peripheral delivery of FNDC5 to the liver leads to elevated blood irisin and increased BDNF expression in the hippocampus. Taken together, this study links endurance exercise and the significant metabolic mediators, PGC-1α and FNDC5, with BDNF expression in the brain [46] (Figure 1).
4. PGC-1α as a Coactivator in WAT Browning, Thermogenesis, and Mitochondrial Biogenesis
Much of the adaptive thermogenesis in small mammals takes place in BAT. BAT is morphologically and metabolically different from WAT and partly exerts opposite physiological functions. Adipocytes from BAT contain multiple small triglyceride-filled droplets as well as a large number of mitochondria. In addition, their mitochondria contain a specific UCP-1, expressed only in brown adipocytes. Genetic studies with mice lacking UCP-1 or PGC-1α in adipocytes indicated that (1) PGC-1α is the only protein that can powerfully activate the UCP-1 enhancer in non-BAT cell lines and (2) when pharmacologically introduced into white adipocytes, PGC-1α induces mitochondrial gene expression and mitochondrial biogenesis. Finally, PGC-1α is a downstream target of adaptive thermogenesis in BAT via adrenergic receptor activation [47,48], the key mechanism in brown-fat differentiation in in vitro cell cultures and in vivo cellular responses to cold exposure [24]. Brown fat and skeletal muscle, in which PGC-1α is highly expressed and can be induced by cold or adrenergic stimuli with enhanced mitochondrial biogenesis, are the two main contributing tissues in adaptive thermogenesis via the adrenergic receptor PGC-1α–UCP-1 axis. Scarpulla and collaborators [49,50] identified and cloned two novel transcription factors, NRF-1 and -2, that bind to the promoter region of the mitochondrial genes β-ATP synthase, cytochrome-c, cytochrome-c-oxidase subunit IV, and mtTFA. PGC-1α has a major effect on the NRF system. When introduced into muscle cells in vitro, PGC-1α greatly induces the gene expression of NRF-1, NRF-2, and mtTFA. Furthermore, PGC-1α interacts directly with NRF-1 and co-activates its transcriptional activity [51] (Figure 1).
5. PGC-1 Controls Cardiac Energy Metabolism in Healthy or Diseased Myocardia
In mammalian embryos, proliferating cardiomyocyte precursor cells rely on glycolysis as their major energy source, and mitochondrial tissue and oxidative metabolism are poorly developed. Once precursor cells differentiate into mature cardiomyocytes, a shift occurs from glycolysis to FA metabolism as the main provider of the entry point for mitochondrial oxidative phosphorylation, which in mature heart cells yields most of the energy [52]. Therefore, during neonatal development, the healthy myocardium increases its rate of β-oxidation while simultaneously decreasing glycolytic activity. Eventually, adult heart muscle derives ~90% of its energy from oxidative phosphorylation in mitochondria, which occupy only ~30% of cardiomyocyte volume [53,54]. During various cardiac disease processes, such as hypertrophy or ischemia-induced cardiomyopathy, both the inhibition of mediators of mitochondrial oxidative phosphorylation (cytochrome-c-oxidase subunits) and the expression or activity of metabolic enzymes involved in oxidative phosphorylation [55,56] were noted [57]. These processes of cardiac remodeling result in a gradual decrease in mitochondrial biogenesis [58], and ATP is utilized for maintaining ion homeostasis rather than for force production during cardiomyocyte contraction; this process leads to irreversible hypertrophy or dilated cardiomyopathy. However, during prolonged periods of cardiac remodeling, cardiomyocyte energy metabolism is regulated by the actions of various transcription factors and their coactivators, such as the PGC-1 family. PGC-1α has been shown to interact with three families of transcription factors: (1) the PPAR family, which regulates the expression of genes involved in FA oxidation; (2) the ERR family; (3) NRF-1 [2,59,60,61], which controls genes that are involved in mitochondrial oxidative phosphorylation and the electron transport chain [62,63]. In cardiomyocytes, PGC-1α is considered a master regulator of metabolism because it co-activates PPARs, ERRs, and NRFs [4,64] and may thereby control the entire metabolic phenotype of cardiomyocytes [7].
In the heart, the interrelationship between PGC-1α and PPARα plays an important role in regulation of the expression of enzymes involved in FAO and uptake pathways [65] and may be involved in regulation of mitochondrial biogenesis [66]. PGC-1α loss of function in murine heart exhibited a damage to mitochondrial respiratory function and reduced expression of genes involved in several mitochondrial metabolic pathways [67,68,69]. Hearts from PGC-1α KO mice showed reductions in mitochondrial enzymatic activities and ATP levels [67]. Arany et al., had shown that PGC-1α KO mice are prone to develop of heart failure in response to transverse aortic constriction (TAC). Furthermore, induction of PGC-1α in cells via catecholamine treatment can reverse the mitochondrial genes inhibition, suggesting that PGC-1α may be a potential therapeutic target in heart failure [68]. In addition, PGC-1α deficient mice cause energy metabolic derangements in multiple systems [69]. Conversely, overexpression of PGC-1α in adult mice had shown a moderate mitochondrial proliferation, abnormal mitochondrial architecture and severe cardiac dysfunction [70], and constitutive overexpression of PGC-1α in murine heart resulted in unconstrained mitochondrial proliferation in cardiac myocytes leading to a dilated cardiomyopathy [10].
Cardiac energy substrate metabolism is disturbed in the hypertrophic and failing heart. The myocardium switches from dependence on fatty acid oxidation (FAO) to glucose utilization in the failing heart, mainly anaerobic glycolysis [71,72,73,74]. These alterations in energy substrate preference are regulated, at least partially, by the downregulation of the genes involved in OXPHOS and FAO and the PPARα–PGC-1α complex [56,73,75,76,77,78]. The expression levels of PPARα and PGC-1α are reduced in several mice models of pressure overload, hypertensive heart disease [68,75,79], ischemic heart disease [57,80,81,82], hypoxia [76], and genetically engineered mouse models of heart failure [83,84,85]. Additionally, under pathologic conditions, PPARα activity is inhibited by the lower levels of the heterodimeric partner, retinoid X receptor (RXR) [86] and by direct phosphorylation, dependent on the extracellular signal-related kinase and mitogen-activated protein kinase (ERK–MAPK) pathway [75]. These findings suggest that deactivation of the cardiac PPARα–PGC-1α axis is an important component of the switch in energy metabolism in the failing heart. It remains to be addressed whether the deactivation of the oxidative metabolism and the PPARα–PGC-1α complex in the hypertrophied and ischemic heart is adaptive or maladaptive. The increment of myocardial reliance on anaerobic glycolytic pathways for ATP production is likely an adaptive response to reduce oxygen consumption. Indeed, partial inhibitors of FAO exhibited a promising therapeutic effect for cardiac disease [87,88,89]. Liao et al., had reported that overexpression of the GLUT1 glucose transporter can prevent pressure overload-induced heart failure [90]. Moreover, overexpression of PGC-1α [83] and PPAR agonists [91,92,93] can prevent cardiac hypertrophy or improve cardiac myocyte contractility.
Cardiovascular disease is extraordinarily widespread in diabetic patients. Cardiomyopathy in diabetic subjects that occurs in the absence of known risk factors (hypertension, hyperlipidemia, etc.) is often referred to as “diabetic cardiomyopathy” [94,95,96,97]. Many studies have proposed that abnormalities in myocardial energy metabolism play an important role in the pathogenesis of diabetic cardiomyopathy. Indeed, the diabetic heart relies nearly exclusively on mitochondrial FAO for ATP requirements [98,99,100,101]. The expression levels of PPARα, PGC-1 α, and various target genes involved in FAO are increased in the murine insulin-resistant [66] and diabetic heart [102,103,104]. Moreover, transgenic mice that overexpress PPARα exclusively in the heart (MHC-PPARα mice) demonstrate a cardiac metabolic phenotype similar to that observed in the diabetic heart, including accelerated rates of FAO, reduction in glucose uptake and utilization, and repression of the mitochondrial biogenic response [66,102]. Mitochondria isolated from diabetic rodents showed reduced rates of OXPHOS [105,106] and decreased efficiency in ATP synthesis [107,108], likely due to increased uncoupled respiration [108]. The importance of PPARs and PGC-1α in the modulation of cardiac energy metabolism makes these regulatory pathways attractive therapeutic targets for diabetic cardiomyopathy.
In summary, increased PPARα and PGC-1α expression with the marked expansion of mitochondrial volume density and oxidative capacity accompany normal cardiac growth during postnatal maturation. Conversely, pathologic hypertrophy is associated with decreased PPARα–PGC-1α expression and/or activity and diminished reliance on oxidative mitochondrial metabolism, which leads to intramyocardial cell lipid accumulation. Finally, gain-of-function studies with PGC-1α overexpression in mice revealed that the extent of cardiomyopathy is primarily determined by the amount of PGC-1α that could be detected in the heart and, more importantly, the moment and duration of its emergence. Thus, both the synthesis and the moment of appearance of PGC-1α play important roles in the regulation of myocardial metabolism and mitochondrial biology (Figure 1).
6. Is PGC-1 a Paracrine Regulator in Epicardial Adipose Tissue?
Recently, a new type of adipose tissue, epicardial adipose tissue (EAT), was found in the heart of patients undergoing open-heart surgery. EAT is physically located next to the myocardium within the lateral wall of the right ventricle and the anterior wall of the left ventricle and surrounds the right coronary and left-anterior descending coronary arteries [109]. Similar to WAT, EAT shows high rates of lipogenesis but also high degrees of WAT lipolysis and thus serves as a local triacylglycerol (TAG) store [110]. EAT contains about five times more UCP-1 mRNA than WAT and also shows high expression of many genes of beige adipose tissue [111], that is, CD137, PRDM16, PGC-1α, C/EBPβ, and PPARα. The present understanding of the potential physiological roles of EAT includes: (1) the release of free FAs as energy to the myocardium under conditions associated with high metabolic demands, (2) the expression of the thermogenic protein UCP-1 in response to cold exposure, and (3) the expression and secretion of specific molecules for cardiovascular protection by vasocrine and paracrine pathways. EAT contributes to cardiovascular protection and vessel remodeling by secreting various paracrine factors. Several EAT-derived factors or cytokines, such as tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), IL-1β, plasminogen activator inhibitor-1 (PAI-1), resistin, and adipokines, have both vasocrine and paracrine effects on the myocardium [112,113]. Other specific molecules secreted from EAT, such as adiponectin and adipocyte-derived relaxing factors called adipokines, can decrease contraction and vasoconstriction by increasing nitric oxide (NO) release or by reducing reactive oxygen species (ROS) production [114]. In addition, macrophages residing in EAT can release anti-inflammatory cytokines such as IL-10 [115]. EAT may contribute to cardioprotection by the local secretion of anti-inflammatory and anti-atherogenic adipokines such as adiponectin and adrenomedullin [116,117]. Both adiponectin and adrenomedullin are directly secreted from EAT into the coronary circulation, and their mRNA levels are correlated with their intracoronary levels [118,119]. Clinically, both adiponectin and adrenomedullin expression in EAT were significantly reduced in patients with coronary artery disease [118,119]. In a clear demonstration of the paracrine regulation of the cardiac function by PGC1 in mice, we found that chronic iron loading attenuated serum adiponectin concentration, thereby resulting in cardiomyopathy. In addition, adiponectin gene (ADIPOQ) overexpression in the heart after adeno-associated virus delivery (AAV-ADIPOQ) ameliorated cardiac iron deposition and restored the cardiac function in iron-overloaded mice; this occurred via the induced expression of heme oxygenase 1 (HO-1) through the PPARα–PGC-1 complex–dependent pathway in cardiomyocytes [120]. Craige et al., created mice with endothelial-specific loss of function (PGC-1α EC KO) that showed significantly reduced PGC-1α expression as well as decreased endothelial NO synthase (eNOS) expression and NO• bioactivity in response to angiotensin-II-induced hypertension [121]. The authors found that PGC-1α EC KO mice had significantly increased blood pressure with vascular dysfunction compared with Cre control mice. In summary, they showed that endothelial PGC-1α expression is required to exert vascular protection via increased bioactivity of NO• through ERRα-induced expression of eNOS, thus preventing cardiovascular disease.
7. Potential for PGC-1α to Modulate Paracrine Regulators in the Heart
PGC-1α is abundantly expressed in tissues with high energy requirements, such as the heart, skeletal muscle, kidney, and BAT [122,123]. In these tissues, PGC-1α controls the expression of genes involved in energy homeostasis, mitochondrial biogenesis, and free FA oxidation function [5,6]. In the heart, cardiomyopathy progression is determined by the amount and the time period of PGC-1α expression. However, the therapeutic window of PGC-1α in cardiomyocytes is relatively narrow because prolonged overexpression of this cofactor leads to uncontrolled mitochondrial proliferation, abnormal sarcomeric structure, and dilated cardiomyopathy [10,124]. Similar phenomena were found in kidney diseases. In the kidney, the basal expression of PGC-1α is stronger in the proximal than the distal tubules, whereas in the glomerulus it is low. A recent study showed aggravated glomerular cell injury when PGC-1 was chronically overexpressed, which is in contrast to the beneficial effects of PGC-1α expression in the proximal tubules promoting acute kidney injury recovery during systemic inflammation [125] or in cisplatin-induced acute renal injury [126]. The cardiac endothelium forms a continuous monolayer of cells that lines the cavity of the heart (endocardial endothelial cells (EECs)) and the luminal surface of the myocardial blood vessels (intramyocardial capillary endothelial cells (IMCEs)). Both EECs and IMCEs can master the contractility of cardiomyocytes by releasing various factors such as NO via endothelial NO-synthase (eNOS), angiotensin II, endothelin-1, peptide growth factors, prostaglandins, and neuregulin-1 (NRG-1) [127]. Craige et al., showed that PGC-1α expression protects the endothelium via increased eNOS expression and NO• bioactivity. ERRα is required for PGC-1α-mediated eNOS expression [121]. Chronic NRG-1 treatment increased oxidative metabolism and mitochondrial activity by enhancing the expression of PGC-1α and PPARδ [128]. Whether PGC-1α can modulate paracrine regulator in the heart needs further investigation. Besides, there are no reports confirming that EAT can act in a paracrine fashion to regulate PGC-1α expression in cardiomyocytes. However, prior reports have indicated that the expression of PGC1α in skeletal muscle may enable the production and release of myokines for the crosstalk between skeletal muscle and other organs. Therefore, future studies should focus on exploring whether PGC-1 in EAT stimulates the secretion of factors that regulate cardiac functions in a paracrine manner, in which cardiac muscle and skeletal muscle can act as endocrine organs.
8. PGC-1α Regulates Metabolic Homeostasis in the Liver
The expression of PGC-1α is induced in the liver at birth [129]. Starvation induces PGC-1α expression in the adult liver via glucagon and glucocorticoid (GR) signaling [130]. The fed-to-fasted transition cause metabolic changes in the liver to promote adaptation to nutrient deprivation. These metabolic changes consist in the activation of hepatic gluconeogenesis, FA β-oxidation, heme biosynthesis, bile acid homeostasis, and synthesis and secretion of ketone bodies [131]. In vitro studies in hepatocytes and in vivo studies have shown that PGC-1α is sufficient to activate the hepatic fasting responses, which include gluconeogenesis, ketogenesis, FA β-oxidation, and bile acid homeostasis [130,132,133]. PGC-1α regulates the metabolic adaption to fasting by coactivating key hepatic transcription factors such as HNF4α, PPARα, GR, FOXO1, LXR, and FXR [4]. PGC-1α-KO mice and RNAi-mediated liver-specific PGC-1α-knockdown mice showed defective gluconeogenic gene expression and hepatic glucose production [134,135]. These mice show a tendency for hypoglycemia and hepatic steatosis upon fasting [69]. In addition, PGC-1α stimulates the expression of genes involved in homocysteine metabolism in cultured primary hepatocytes and in the liver [136]. Hepatic PGC-1α protein expression and activation of mitochondrial biogenesis were reduced in a mouse model of hepatic steatosis [137]. PGC-1α plays an important role in exercise-induced hepatic mitochondrial adaptation [138]. PGC-1α expression was lower in the liver of obese, sedentary humans than lean humans [139]. Overexpression of hepatic PGC-1α increased hepatic FA oxidation with decreased TAG storage and secretion in vivo and in vitro [140]. In addition, PGC-1α integrates the mammalian clock and energy metabolism. PGC-1α stimulates the gene expression of the clock genes Bmal1 (Arntl) and Rev-erbα (Nr1d1) by coactivation of the receptor tyrosine kinase-like orphan receptor family of orphan nuclear receptors. PGC-1α-null mice show abnormal diurnal rhythms of activity, body temperature, and metabolic rate [141]. Therefore, PGC-1α regulates both the fed-to-fasted energy transition and the diurnal rhythm in liver metabolic homeostasis.
9. PGC-1α Regulates Kidney Metabolism via Mitochondrial Homeostasis
As a bridge between homeostasis and mitochondrial function, PGC-1α activates NRF-1 and -2, which are nuclear-encoded transcription factors that promote the expression of multiple genes involved in mitochondrial DNA transcription and mitochondrial respiratory chains with anti-oxidative effects [142,143]. In the kidney, PGC-1α is predominantly expressed in the proximal tubules, and enforced expression of PGC-1α in cultured proximal tubular cells increased mitochondrial number, respiratory capacity, and mitochondrial protein level, which indicates the effectiveness of PGC-1α in proximal tubular homeostasis [122,144]. In the septic acute kidney injury (AKI) model, PGC-1α expression in tubular cells was proportionally decreased with an increasing degree of renal impairment. Although mice with PGC-1α gene deletion do not show altered kidney size [69,134], they exhibit increased serum blood urea nitrogen (BUN) and creatinine levels in these models [125], and patients and mouse models with acute and chronic kidney disease commonly show decreased PGC-1α expression accompanied by reduced FA oxidation. In addition, treatment with the PPARγ agonist rosiglitazone could induce PGC-1α expression in the nucleus of renal mesangial cells and significantly ameliorate renal fibrosis in mouse models of diabetic kidney disease. Furthermore, in vitro experiments with cultured renal mesangial cells demonstrated that PGC-1α knockdown increased glucose-induced ROS levels [145].
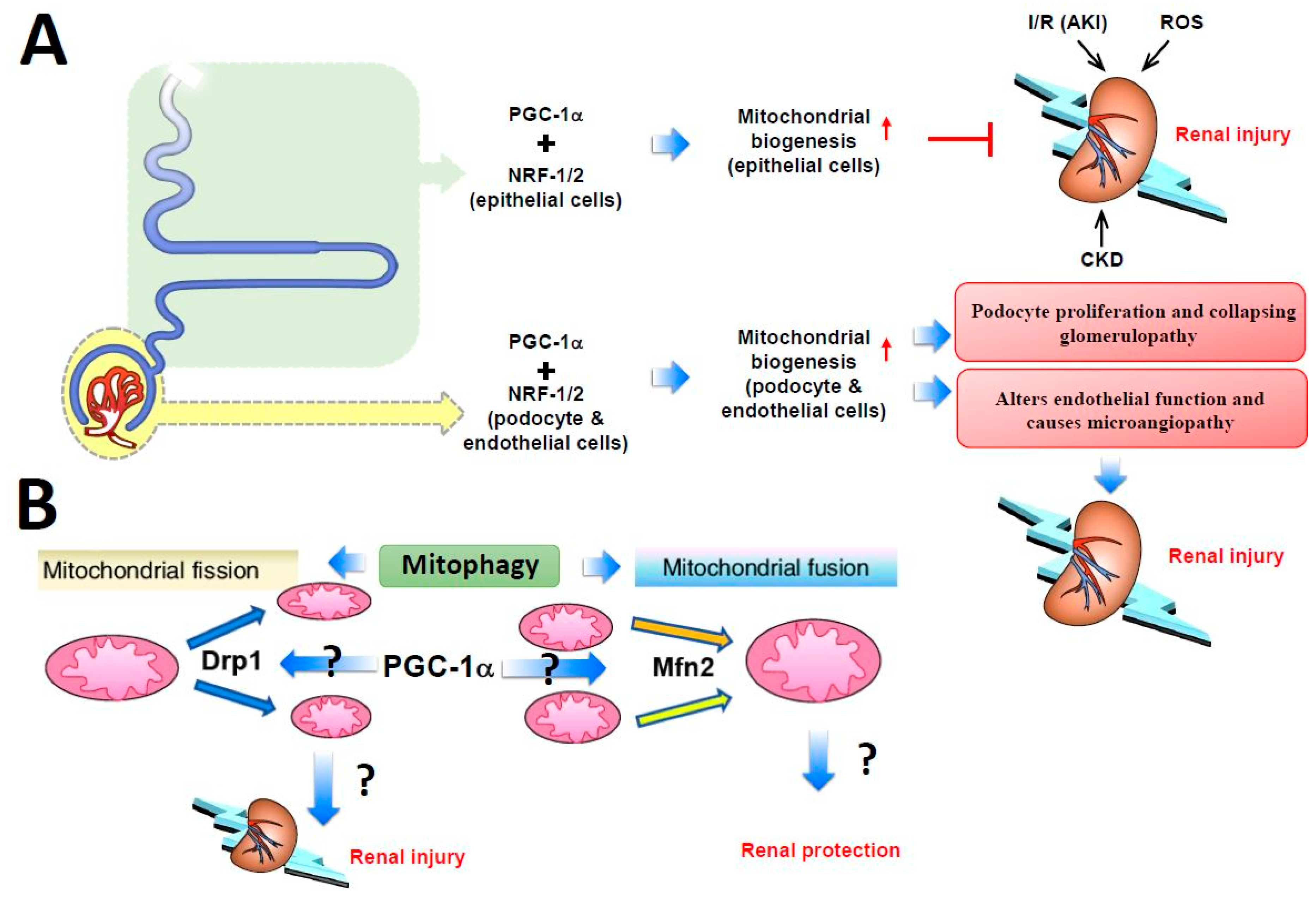
Studies from Rasbach et al., who used tertbutyl hydroperoxide (tBHP), an agent that profoundly depletes cellular glutathione, to induce oxidative stress in the rabbit proximal tubular cell culture system resulted in iron-dependent lipid peroxidation with extensive primary mitochondrial damage [146]. PGC-1α protein level was greatly increased after tBHP treatment, and the increase could be blocked by inhibiting the epidermal growth factor receptor–Src–p38 MAPK axis pathway. However, adenovirus-induced PGC-1α overexpression produced a 25% to 50% increase in mitochondrial number [147], which had a protective effect against tBHP-induced cell damage [144]. Choi et al., found that PGC1-α could attenuate ischemia-reperfusion-induced acute kidney injury by ameliorating the mitochondria dysfunction mediated by p38 signaling [148]. These consistent in vivo and in vitro findings indicate that PGC-1α expression may be increased in the early stage of acute and chronic kidney injury as a compensatory response and PGC-1α can regulate renal tubular mitochondrial biogenesis. Other kidney cells, such as podocytes and endothelial cells, are less metabolically active and have a narrow PGC-1α tolerance. Increasing PGC-1α levels in podocytes induce podocyte proliferation and collapsing glomerulopathy development, whereas increasing PGC-1α levels in endothelial cells alter the endothelial function and cause microangiopathy, thus highlighting the cell type-specific role of PGC-1α in the kidney (Figure 2A).
Defects in mitochondrial fusion–fission homeostasis lead to altered mitochondrial morphology and impaired mitochondrial function and cause tubular damage in acute kidney injury. In addition, the balance between mitochondrial fusion and fission shifts to mitochondrial fission, resulting in mitochondrial fragmentation and then altered mitochondrial structure and renal tubular cell apoptosis [149]. Brooks et al. [149] observed mitochondrial fragmentation and Drp1 mobilization to the outer mitochondrial membrane in injured tubular cells. Drp1, a mitochondrial fission mediator, is activated rapidly after ischemia-reperfusion-induced injury and induces mitochondrial fragmentation and subsequent renal tubular cell apoptosis [149]. By using dominant-negative mutants and RNA interference, Jiang et al., demonstrated that Drp1 inhibition attenuated mitochondrial fragmentation, preserved mitochondrial integrity, limited renal cell apoptosis, and preserved kidney function. However, pharmacological inhibition or genetic deletion of autophagy-related genes worsened renal injury. These inconsistent results may imply that excessive mitochondrial fission during acute kidney injury is deleterious to organ function, and safe clearance of damaged mitochondria via mitophagy may be protective [150,151]. Meanwhile, primary cultured cells with tissue-specific knockout of Mfn2, a mitochondrial fusion mediator, in renal proximal tubular cells were highly sensitive to Bax activation and cytochrome c release, which led to cell apoptosis. However, Mfn2 is also known to suppress cell proliferative effects via p21Ras, independently of mitochondrial dynamics [152,153]. Such Mfn2-mediated hyperplasia suppression may contribute greatly to renal recovery after stress; therefore, reducing Mfn2 level in proximal tubular cells might actually accelerate organ recovery [154,155]. Gall et al., had shown that conditional knockout of proximal tubule Mfn2 markedly boosts recovery of renal function and increased rodent survival after acute renal ischemia, partially by activating Ras and ERK1/2 signaling [156]. The above findings indicate, in general, that increased Drp1 or decreased Mfn2 levels exacerbate renal tubular damage via an imbalance in mitochondrial fission and fusion, with subsequent enhancement of mitochondrial fragmentation and aggravated acute kidney injury; however, studies with opposite results were also reported (Figure 2B). Further research is needed to investigate whether PGC-1α evokes the performance of the mitochondrial genes via the Drp1–Mfn2 balance pathway and thus affects the function of the kidney in health or disease.
10. PGC-1α Regulates Cancer Metabolism
Metabolic reprogramming occurring in cancer cells refers to the ability to grow and survive under nutrient-starved or stressful microenvironments [157,158]. Increments in glycolysis, glutaminolytic flux, amino acid and lipid metabolism, and mitochondrial biogenesis have been observed in cancer development [159,160,161,162]. Deregulated metabolism is associated with oncogenesis, including the phenomenon of epithelial-to-mesenchymal transition (EMT), a complicated process that enables cancer cells to invade neighboring tissues and migrate to the vasculature [163,164,165]. Among the numerous regulators of cancer metabolism, PGC-1α has been shown to regulate many processes linked to oncogenesis by, for example, promoting the expression of antioxidant genes which protect cells from the detrimental effects of ROS, enhancing the catabolism of glucose and fatty acids, and promoting gluconeogenesis and lipogenesis which perform opposite anabolic functions [166,167,168,169,170]. No specific variant or isoform of PGC-1α has been reported in cancer studies. Some studies have shown that biphasic expression of PGC1α was observed in cancer biopsies or cells of breast cancer [171,172,173,174], melanoma [175,176,177], colon cancer [169,178], and ovarian cancer [179,180,181]. Low PGC-1α levels are associated with a worse outcome in breast and liver carcinomas [171,172,182]. The chemoresistant clear-cell subtype of ovarian carcinoma was identified by the lack of expression of both PGC-1α and mitochondrial transcription factor A (TFAM) [180]. In contrast, some studies showed that the plasma concentrations of PGC1α in breast cancer patients were higher than in healthy groups, and a multivariate analysis showed a correlation between high levels of PGC-1α and worse prognosis [183]. In a report of prostate cancer, androgens signaling via AMPK caused the increment of PGC1α mitobiogenesis, OXPHOS, and glycolysis. Furthermore, findings in mouse xenografts and patient samples suggested that AMPK–PGC1α function was associated with prostate cancer growth [184].
Even though many studies have investigated the role of PGC-1α in cancer by examining its expression via PGC-1α overexpression and siRNA knockdown experiments, the role of PGC-1α in cancer is still controversial. Several studies have shown that PGC-1α has tumor-suppressive effects. PGC-1α overexpression in melanoma cells by adenovirus infection suppressed metastasis via the direct regulation of inhibitor of DNA binding protein 2 (ID2) and the inhibition of transcription factor 4 (TCF4)-mediated gene transcription [177]. Human ovarian cancer cell line Ho-8910 overexpressing PGC-1α has been shown to undergo apoptosis through downregulation of B-cell lymphoma 2 (Bcl-2) and upregulation of Bcl2-associated X protein (Bax) [185]. Wang et al., revealed that increased PGC-1α expression by a PPAR pan-agonist (bezafibrate) upregulated mitochondrial biogenesis, resulting in the inhibition of proliferation and invasion in HeLa, 143B, and MDA-MB-231 cancer cells [186]. Overexpression of PGC-1α by adenovirus infection in HepG2 human hepatoma cells upregulated E-cadherin expression and inhibited cell motility [187]. A study by Torrano et al., showed that PGC-1α inhibited the metastasization of prostate carcinoma via an estrogen-related receptor alpha (ERRα)-dependent transcriptional program [188]. PGC-1α overexpression in HT29 and HCT116 colorectal cancer cells induced apoptosis through ROS accumulation [178].
As opposed to the tumor-suppressive role of PGC-1α described above, many reports have shown that PGC-1α is a tumor promoter [169,170,176,184,189,190,191]. Bhalla et al., demonstrated that PGC-1α knockout mice had reduced chemical-induced liver and colon carcinogenesis, suggesting that PGC-1α may induce carcinogenesis [169]. This study reported that PGC-1α stimulates carcinogenesis and tumor growth via the induction of lipogenic enzymes (fatty acid synthase and acetyl-CoA carboxylase) in genetically modified PGC-1α mice [169]. In addition, knockdown PGC-1α significantly induced apoptosis in PGC-1α-positive melanoma cell lines, suggesting that PGC-1α regulates the survival of PGC-1α-positive melanoma cells [176]. PGC-1α promoted prostate cancer cell growth through the activation of androgen receptor [184,189]. It was shown that cell proliferation was inhibited in PGC-1α siRNA knockdown experiments in H1944 lung adenocarcinoma cells [191]. Similarly, overexpression of PGC-1α induced HEK293 cell proliferation and tumorigenesis through the upregulation of Specificity protein 1 (Sp1) and acyl-CoA-binding protein [190]. PGC-1α overexpression or ERRα activation conferred breast cancer cell growth ability, even under hypoxia conditions [170]. Despite the fact that PGC-1α can act as a tumor suppressor and a tumor promoter, there is no explicitly defined mechanism that can explain its dichotomous effects. However, its dual actions can be partially explained by its cell type-specific effects and varied interacting proteins.
11. Conclusions
PPARα and PGC-1α play a central role in metabolic flexibility by driving robust and coordinated changes in the expression of key components of mitochondrial biogenesis and by performing a critical metabolic regulation in many vital organs, including adipose tissue, skeletal muscle, heart, liver, and kidney (Figure 1). Traditionally, in BAT and WAT, mitochondrial biogenesis and BAT gene expression are regulated by PGC-1α. Adrenergic stimulation and reduced temperature trigger signaling cascades including the upregulation of UCP-1 level, thereby resulting in body thermogenesis. In skeletal muscle and WAT, the transcriptional activity of PGC-1α is responsible for the expression of gene networks that control glucose uptake, glycolysis, FA oxidation, the TCA cycle, OXPHOS, and mitochondrial biogenesis and uncoupling. Therefore, increased exercise will increase mitochondrial gene biogenesis and the secretion of myokines (such as irisin), resulting in WAT browning and liver gluconeogenesis and preventing obesity and insulin resistance. In EAT, increased HO-1 expression depends on the PGC-1α–UCP-1 axis, which subsequently decreases free radical and ROS production, thus reducing cardiomyopathy. However, whether long-term PGC-1α overexpression improves or impairs heart or kidney function under disease- or stress-induced remodeling is unclear. In cancer, the dichotomous effects of PGC-1α can be partially explained by its cell type-specific effects and diverse interacting proteins. Therefore, more details in vivo and pre-clinical work are required to assess the usefulness of PGC-1α-inducing drugs in cardiovascular, renal, and cancer therapy.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Besseiche, A.; Riveline, J.P.; Gautier, J.F.; Breant, B.; Blondeau, B. Metabolic roles of PGC-1alpha and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Popov, D.V.; Lysenko, E.A.; Kuzmin, I.V.; Vinogradova, V.; Grigoriev, A.I. Regulation of PGC-1alpha Isoform Expression in Skeletal Muscles. Acta Nat. 2015, 7, 48–59. [Google Scholar]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatazawa, Y.; Senoo, N.; Tadaishi, M.; Ogawa, Y.; Ezaki, O.; Kamei, Y.; Miura, S. Metabolomic Analysis of the Skeletal Muscle of Mice Overexpressing PGC-1alpha. PLoS ONE 2015, 10, e0129084. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.A.; Daniels, T.G.; Wang, X.; Paul, A.; Lin, J.; Spiegelman, B.M.; Stevenson, S.C.; Rangwala, S.M. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 2008, 104, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, D.O.; Boros, L.G.; Crunkhorn, S.; Gami, H.; Patti, M.E. Dual modulation of both lipid oxidation and synthesis by peroxisome proliferator-activated receptor-gamma coactivator-1alpha and -1beta in cultured myotubes. FASEB J. 2010, 24, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Mukai, K.; Lally, J.S.; Maher, A.C.; Gurd, B.J.; Heigenhauser, G.J.; Spriet, L.L.; Holloway, G.P. AMP-activated protein kinase is required for exercise-induced peroxisome proliferator-activated receptor co-activator 1 translocation to subsarcolemmal mitochondria in skeletal muscle. J. Physiol. 2013, 591, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Zheng, D.; Houmard, J.A.; Brault, J.J.; Hickner, R.C.; Cortright, R.N. Overexpression of PGC-1alpha increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E253–E263. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Baum, O.; Santos, G.; Hoppeler, H.; Handschin, C. Peroxisome proliferator-activated receptor {gamma} coactivator 1{alpha} (PGC-1{alpha}) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Boil. Chem. 2010, 285, 32793–32800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supruniuk, E.; Miklosz, A.; Chabowski, A. The Implication of PGC-1alpha on Fatty Acid Transport across Plasma and Mitochondrial Membranes in the Insulin Sensitive Tissues. Front. Physiol. 2017, 8, 923. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W.; Thomason, D.B. Molecular and cellular adaptation of muscle in response to exercise: Perspectives of various models. Physiol. Rev. 1991, 71, 541–585. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 2001, 90, 1137–1157. [Google Scholar] [CrossRef] [PubMed]
- Bassel-Duby, R.; Olson, E.N. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Thom, R.; Rowe, G.C.; Jang, C.; Safdar, A.; Arany, Z. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor gamma coactivator (PGC)-1alpha. J. Boil. Chem. 2014, 289, 8810–8817. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Boil. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Gupta, R.K.; Ruas, J.L.; Wu, J.; Naseri, E.; Estall, J.L.; Spiegelman, B.M. PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc. Natl. Acad. Sci. USA 2010, 107, 21866–21871. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.Y. The role of exercise-induced myokines in regulating metabolism. Arch. Pharm. Res. 2018, 41, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskalopoulou, S.S.; Cooke, A.B.; Gomez, Y.H.; Mutter, A.F.; Filippaios, A.; Mesfum, E.T.; Mantzoros, C.S. Plasma irisin levels progressively increase in response to increasing exercise workloads in young, healthy, active subjects. Eur. J. Endocrinol. 2014, 171, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.Y.; Mougios, V.; Kabasakalis, A.; Fatouros, I.; Siopi, A.; Douroudos, I.I.; Filippaios, A.; Panagiotou, G.; Park, K.H.; Mantzoros, C.S. Exercise-induced irisin secretion is independent of age or fitness level and increased irisin may directly modulate muscle metabolism through AMPK activation. J. Clin. Endocrinol. Metab. 2014, 99, E2154–E2161. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Linderman, J.D.; Smith, S.; Brychta, R.J.; Wang, J.; Idelson, C.; Perron, R.M.; Werner, C.D.; Phan, G.Q.; Kammula, U.S.; et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014, 19, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Becerril, S.; Mendez-Gimenez, L.; Ramirez, B.; Sainz, N.; Catalan, V.; Gomez-Ambrosi, J.; Fruhbeck, G. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 2015, 39, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Dincer, F.; Mesfum, E.; Mantzoros, C.S. Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int. J. Obes. 2014, 38, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, R.; Jiang, F.; Wang, J.; Chen, M.; Peng, D.; Yan, J.; Wang, S.; Bao, Y.; Hu, C.; et al. Circulating irisin levels are associated with lipid and uric acid metabolism in a Chinese population. Clin. Exp. Pharmacol. Physiol. 2015, 42, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.L.; Hittel, D.S.; McPherron, A.C. Expression and function of myostatin in obesity, diabetes, and exercise adaptation. Med. Sci. Sports Exerc. 2011, 43, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Streeper, R.S.; Farese, R.V., Jr.; Yamamoto, K.R. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc. Natl. Acad. Sci. USA 2006, 103, 15675–15680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wall, R.J.; Yang, J. Transgenic expression of myostatin propeptide prevents diet-induced obesity and insulin resistance. Biochem. Biophys. Res. Commun. 2005, 337, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2012, 55, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Dong, Y.; Dong, Y.; Chen, F.; Mitch, W.E.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Liang, X.; Bi, P.; Kuang, S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1alpha-Fndc5 pathway in muscle. FASEB J. 2013, 27, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Hefti, F. BDNF and NGF treatment in lesioned rats: Effects on cholinergic function and weight gain. Neuroreport 1992, 3, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Choi, E.Y.; Liu, X.; Martin, A.; Wang, C.; Xu, X.; During, M.J. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 2011, 14, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Jacobsson, A.; Rehnmark, S.; Nedergaard, J. Signal transduction in brown adipose tissue recruitment: Noradrenaline and beyond. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1996, 20 (Suppl. 3), S36–S42. [Google Scholar]
- Ricquier, D. Molecular biology of brown adipose tissue. Proc. Nutr. Soc. 1989, 48, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Boil. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Huang, H.; McMullen, M.R.; Mandal, P.; Sun, L.; Nagy, L.E. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J. Boil. Chem. 2008, 283, 26850–26858. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Dudycha, S.J.; O’Rourke, B. Cardiac energy metabolism: Models of cellular respiration. Annu. Rev. Biomed. Eng. 2001, 3, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol. 2004, 555 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Delomenie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551 Pt 2, 491–501. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Boil. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Tuomainen, T.; Tavi, P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp. Cell Res. 2017, 360, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Boil. 2000, 20, 1868–1876. [Google Scholar] [CrossRef]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Boil. 2005, 3, e101. [Google Scholar]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004, 94, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267 Pt 2, H742–H750. [Google Scholar] [CrossRef]
- Christe, M.E.; Rodgers, R.L. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J. Mol. Cell. Cardiol. 1994, 26, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Overturf, M.L. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 1988, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Massie, B.M.; Schaefer, S.; Garcia, J.; McKirnan, M.D.; Schwartz, G.G.; Wisneski, J.A.; Weiner, M.W.; White, F.C. Myocardial high-energy phosphate and substrate metabolism in swine with moderate left ventricular hypertrophy. Circulation 1995, 91, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Razeghi, P.; Essop, M.F.; Huss, J.M.; Abbasi, S.; Manga, N.; Taegtmeyer, H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem. Biophys. Res. Commun. 2003, 303, 1024–1027. [Google Scholar] [CrossRef]
- Van Bilsen, M.; Smeets, P.J.; Gilde, A.J.; van der Vusse, G.J. Metabolic remodelling of the failing heart: The cardiac burn-out syndrome? Cardiovasc. Res. 2004, 61, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Van Bilsen, M. “Energenetics” of heart failure. Ann. N. Y. Acad. Sci. 2004, 1015, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Laws, F.A.; Goodwin, G.W.; Taegtmeyer, H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J. Boil. Chem. 2001, 276, 44390–44395. [Google Scholar] [CrossRef] [PubMed]
- Remondino, A.; Rosenblatt-Velin, N.; Montessuit, C.; Tardy, I.; Papageorgiou, I.; Dorsaz, P.A.; Jorge-Costa, M.; Lerch, R. Altered expression of proteins of metabolic regulation during remodeling of the left ventricle after myocardial infarction. J. Mol. Cell. Cardiol. 2000, 32, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt-Velin, N.; Montessuit, C.; Papageorgiou, I.; Terrand, J.; Lerch, R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc. Res. 2001, 52, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Sharma, S.; Adrogue, J.; Salazar, R.; Duerr, G.D.; Crapo, J.D.; Entman, M.L.; Taegtmeyer, H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation 2005, 112, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Wang, S.C.; Shirai, M.; Scaglia, F.; Xie, M.; Sakai, S.; Tanaka, T.; Kulkarni, P.A.; Barger, P.M.; Youker, K.A.; et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004, 23, 3559–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiguchi, K.; Tian, Q.; Ishiyama, M.; Burchfield, J.; Gao, F.; Mann, D.L.; Barger, P.M. Inhibition of PPAR-alpha activity in mice with cardiac-restricted expression of tumor necrosis factor: Potential role of TGF-beta/Smad3. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1443–H1451. [Google Scholar] [CrossRef] [PubMed]
- Pellieux, C.; Aasum, E.; Larsen, T.S.; Montessuit, C.; Papageorgiou, I.; Pedrazzini, T.; Lerch, R. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J. Mol. Cell. Cardiol. 2006, 41, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Levy, F.H.; Kelly, D.P. Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: A mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J. Boil. Chem. 2001, 276, 27605–27612. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R.S.; Hill, J.A. The use of ranolazine in cardiovascular disease. Expert Opin. Investig. Drugs 2002, 11, 117–123. [Google Scholar] [PubMed]
- Rupp, H.; Zarain-Herzberg, A.; Maisch, B. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002, 27, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.P.; Stanley, W.C.; Morita, H.; Suzuki, G.; Roth, B.A.; Blackburn, B.; Wolff, A.; Sabbah, H.N. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ. Res. 2002, 91, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Jain, M.; Cui, L.; D’Agostino, J.; Aiello, F.; Luptak, I.; Ngoy, S.; Mortensen, R.M.; Tian, R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002, 106, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Takano, H.; Nagai, T.; Uozumi, H.; Hasegawa, H.; Kubota, N.; Saito, T.; Masuda, Y.; Kadowaki, T.; Komuro, I. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation 2002, 105, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ohki, R.; Lee, R.T.; Ikeda, U.; Shimada, K. Peroxisome proliferator-activated receptor gamma activators inhibit cardiac hypertrophy in cardiac myocytes. Circulation 2001, 104, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Rodriguez-Calvo, R.; Jove, M.; Michalik, L.; Wahli, W.; Laguna, J.C.; Vazquez-Carrera, M. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005, 65, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Fein, F.S.; Sonnenblick, E.H. Diabetic cardiomyopathy. Cardiovasc. Drugs Ther. 1994, 8, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [PubMed]
- Keen, H.; Jarrett, R.J. The WHO multinational study of vascular disease in diabetes: 2. Macrovascular disease prevalence. Diabetes Care 1979, 2, 187–195. [Google Scholar] [PubMed]
- Fein, F.S.; Sonnenblick, E.H. Diabetic cardiomyopathy. Prog. Cardiovasc. Dis. 1985, 27, 255–270. [Google Scholar] [CrossRef]
- Gamble, J.; Lopaschuk, G.D. Glycolysis and glucose oxidation during reperfusion of ischemic hearts from diabetic rats. Biochim. Biophys. Acta 1994, 1225, 191–199. [Google Scholar] [CrossRef]
- Stanley, W.C.; Lopaschuk, G.D.; McCormack, J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 1997, 34, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.; McNeill, J.H. The diabetic heart: Metabolic causes for the development of a cardiomyopathy. Cardiovasc. Res. 1992, 26, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, C.; Weng, S.; Feng, C.; Finck, B.N.; Knutsen, R.H.; Leone, T.C.; Coleman, T.; Mecham, R.P.; Kelly, D.P.; Semenkovich, C.F. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat. Med. 2003, 9, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.H.; Moore, K.H.; Giacomelli, F.; Wiener, J. Defective oxidative metabolism of heart mitochondria from genetically diabetic mice. Diabetes 1983, 32, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Konno, N.; Kako, K.J. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc. Res. 1992, 26, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Epicardial and pericardial fat: Close, but very different. Obesity 2009, 17, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, T.P.; Czech, M.P. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: Basic mechanisms and clinical associations. J. Am. Heart Assoc. 2014, 3, e000582. [Google Scholar] [CrossRef] [PubMed]
- Sacks, H.S.; Fain, J.N.; Holman, B.; Cheema, P.; Chary, A.; Parks, F.; Karas, J.; Optican, R.; Bahouth, S.W.; Garrett, E.; et al. Uncoupling protein-1 and related messenger ribonucleic acids in human epicardial and other adipose tissues: Epicardial fat functioning as brown fat. J. Clin. Endocrinol. Metab. 2009, 94, 3611–3615. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Lopaschuk, G.D.; Carvalho, E. Cellular cross-talk between epicardial adipose tissue and myocardium in relation to the pathogenesis of cardiovascular disease. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E937–E949. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldine, W.; Ghantous, C.M.; Zibara, K.; Dib, L.; Issaa, H.; Itani, H.A.; El-Zein, N.; Zeidan, A. Adiponectin Attenuates Angiotensin II-Induced Vascular Smooth Muscle Cell Remodeling through Nitric Oxide and the RhoA/ROCK Pathway. Front. Pharmacol. 2016, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.; Ahswin, H.; Smart, N.; Bayon, Y.; Wohlert, S.; Hunt, J.A. Reactive oxygen species (ROS)—A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur. Cells Mater. 2012, 24, 249–265. [Google Scholar] [CrossRef]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; di Gioia, C.R. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with coronary artery disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Silaghi, A.; Achard, V.; Paulmyer-Lacroix, O.; Scridon, T.; Tassistro, V.; Duncea, I.; Clement, K.; Dutour, A.; Grino, M. Expression of adrenomedullin in human epicardial adipose tissue: Role of coronary status. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1443–E1450. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Bianco, A.C. Epicardial adipose tissue: Emerging physiological, pathophysiological and clinical features. Trends Endocrinol. Metab. TEM 2011, 22, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; di Gioia, C.R.; Cotesta, D.; Petramala, L.; Travaglini, C.; De Santis, V.; Vitale, D.; Tritapepe, L.; Letizia, C. Epicardial adipose tissue adiponectin expression is related to intracoronary adiponectin levels. Horm. Metab. Res. 2009, 41, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lian, W.S.; Chen, H.H.; Lai, P.F.; Cheng, C.F. Adiponectin ameliorates iron-overload cardiomyopathy through the PPARalpha-PGC-1-dependent signaling pathway. Mol. Pharmacol. 2013, 84, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kroller-Schon, S.; Li, C.; Kant, S.; Cai, S.; Chen, K.; Contractor, M.M.; Pei, Y.; Schulz, E.; Keaney, J.F., Jr. PGC-1alpha dictates endothelial function through regulation of eNOS expression. Sci. Rep. 2016, 6, 38210. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining ancient organelles: Mitochondrial biogenesis and maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Noireaud, J.; Andriantsitohaina, R. Recent insights in the paracrine modulation of cardiomyocyte contractility by cardiac endothelial cells. BioMed Res. Int. 2014, 2014, 923805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Pich, S.; Paz, J.C.; Sanches, R.; Martinez, V.; Orpinell, M.; Palacin, M.; Zorzano, A.; Guma, A. Neuregulins increase mitochondrial oxidative capacity and insulin sensitivity in skeletal muscle cells. Diabetes 2007, 56, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tarr, P.T.; Yang, R.; Rhee, J.; Puigserver, P.; Newgard, C.B.; Spiegelman, B.M. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Boil. Chem. 2003, 278, 30843–30848. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): Requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Arning, E.; Liu, C.; Vitvitsky, V.; Hernandez, C.; Banerjee, R.; Bottiglieri, T.; Lin, J.D. Regulation of homocysteine homeostasis through the transcriptional coactivator PGC-1alpha. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E543–E548. [Google Scholar] [CrossRef] [PubMed]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Haase, T.N.; Ringholm, S.; Leick, L.; Bienso, R.S.; Kiilerich, K.; Johansen, S.; Nielsen, M.M.; Wojtaszewski, J.F.; Hidalgo, J.; Pedersen, P.A.; et al. Role of PGC-1alpha in exercise and fasting-induced adaptations in mouse liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1501–R1509. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.A.; Eagon, J.C.; LaRiviere, L.L.; Korenblat, K.M.; Klein, S.; Finck, B.N. Hepatic lipin 1beta expression is diminished in insulin-resistant obese subjects and is reactivated by marked weight loss. Diabetes 2007, 56, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Meers, G.M.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Schnellmann, R.G. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 2007, 355, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [PubMed]
- Sogabe, K.; Roeser, N.F.; Venkatachalam, M.A.; Weinberg, J.M. Differential cytoprotection by glycine against oxidant damage to proximal tubule cells. Kidney Int. 1996, 50, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Schnellmann, R.G. Signaling of mitochondrial biogenesis following oxidant injury. J. Boil. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1alpha attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3beta inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.; Cho, S.G.; Wang, C.Y.; Yang, T.; Dong, Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C447–C455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.P.; et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Boil. 2004, 6, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 2014, 28, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, J.K.; Harris, R.C. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 2012, 82, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Han, S.J.; Kim, J.I.; Lee, S.; Lipschutz, J.H.; Park, K.M. Activation of ERK accelerates repair of renal tubular epithelial cells, whereas it inhibits progression of fibrosis following ischemia/reperfusion injury. Biochim. Biophys. Acta 2013, 1832, 1998–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, J.M.; Wang, Z.; Bonegio, R.G.; Havasi, A.; Liesa, M.; Vemula, P.; Borkan, S.C. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J. Am. Soc. Nephrol. JASN 2015, 26, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Boil. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. TIG 2017, 33, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Watkins, G.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. The localisation and reduction of nuclear staining of PPARgamma and PGC-1 in human breast cancer. Oncol. Rep. 2004, 12, 483–488. [Google Scholar] [PubMed]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Boil. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabaries, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chenard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, I.; Salvatore, L.; Murzilli, S.; Lo Sasso, G.; Latorre, D.; Martelli, N.; Egorova, A.V.; Polishuck, R.; Madeyski-Bengtson, K.; Lelliott, C.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. USA 2011, 108, 6603–6608. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielson, M.; Bjorklund, M.; Carlson, J.; Shoshan, M. Expression of mitochondrial regulators PGC1alpha and TFAM as putative markers of subtype and chemoresistance in epithelial ovarian carcinoma. PLoS ONE 2014, 9, e107109. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1alpha induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [PubMed]
- Liu, R.; Zhang, H.; Zhang, Y.; Li, S.; Wang, X.; Wang, X.; Wang, C.; Liu, B.; Zen, K.; Zhang, C.Y.; et al. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha acts as a tumor suppressor in hepatocellular carcinoma. Tumour Boil. 2017, 39, 1010428317695031. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.F.; Xu, C.; Pan, X.; Cai, L.; Lin, X.Y.; Chen, S.; Biskup, E. Prognostic value of plasma levels of HIF-1a and PGC-1a in breast cancer. Oncotarget 2016, 7, 77793–77806. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ba, Y.; Liu, C.; Sun, G.; Ding, L.; Gao, S.; Hao, J.; Yu, Z.; Zhang, J.; Zen, K.; et al. PGC-1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma-dependent pathway. Cell Res. 2007, 17, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Su, Y.; Yin, P.H.; Lee, H.C.; Chi, C.W. PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer. Res. 2009, 29, 5057–5063. [Google Scholar] [PubMed]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Boil. 2016, 18, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Fujimoto, N.; Seki, N.; Naito, S. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol. Endocrinol. 2010, 24, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Yun, S.H.; Park, E.S.; Jeong, J.S.; Kwak, J.Y.; Park, J.I. Overexpression of PGC1alpha enhances cell proliferation and tumorigenesis of HEK293 cells through the upregulation of Sp1 and Acyl-CoA binding protein. Int. J. Oncol. 2015, 46, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Delgado, O.; Celiktas, M.; Katayama, H.; Wang, H.; Gazdar, A.F.; Hanash, S.M. Proteomic signatures associated with p53 mutational status in lung adenocarcinoma. Proteomics 2014, 14, 2750–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic description of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) function in various organs. Traditionally, in brown adipocytes (BAT) and white adipocytes (WAT), mitochondrial biogenesis and BAT gene expression are regulated by PGC-1α. Adrenergic stimulation and lower temperature trigger signaling cascades, including the upregulation of UCP-1 level, thereby resulting in body thermogenesis. In skeletal muscle and WAT, the transcriptional activity of PGC-1α is responsible for the expression of gene networks that control glucose uptake, glycolysis, fatty acid (FA) oxidation, tricarboxylic acid cycle, oxidative phosphorylation (OXPHOS), mitochondrial biogenesis, and protein uncoupling. Therefore, increasing exercise will increase mitochondrial gene biogenesis and secretion of myokines (such as irisin), which results in WAT browning and liver gluconeogenesis to prevent obesity and insulin resistance. In epicardial adipose tissue (EAT), increased heme oxygenase 1 (HO-1) expression depends on the PGC-1α–UCP-1 axis activity, which then decreases free radicals and reactive oxygen species (ROS) production, thus reducing cardiomyopathy. However, whether increased expression of cytokines such as TNF-α, IL-6, or adipokines by the PGC-1α–UCP-1 axis can reduce cardiomyopathy or not is still unclear.
Figure 1.
Schematic description of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) function in various organs. Traditionally, in brown adipocytes (BAT) and white adipocytes (WAT), mitochondrial biogenesis and BAT gene expression are regulated by PGC-1α. Adrenergic stimulation and lower temperature trigger signaling cascades, including the upregulation of UCP-1 level, thereby resulting in body thermogenesis. In skeletal muscle and WAT, the transcriptional activity of PGC-1α is responsible for the expression of gene networks that control glucose uptake, glycolysis, fatty acid (FA) oxidation, tricarboxylic acid cycle, oxidative phosphorylation (OXPHOS), mitochondrial biogenesis, and protein uncoupling. Therefore, increasing exercise will increase mitochondrial gene biogenesis and secretion of myokines (such as irisin), which results in WAT browning and liver gluconeogenesis to prevent obesity and insulin resistance. In epicardial adipose tissue (EAT), increased heme oxygenase 1 (HO-1) expression depends on the PGC-1α–UCP-1 axis activity, which then decreases free radicals and reactive oxygen species (ROS) production, thus reducing cardiomyopathy. However, whether increased expression of cytokines such as TNF-α, IL-6, or adipokines by the PGC-1α–UCP-1 axis can reduce cardiomyopathy or not is still unclear.

Figure 2.
Schematic description of PGC-1α function in renal homeostasis. (A) PGC-1α associated with nuclear respiratory factors 1 and 2 (NRF-1/2) has a protective role in renal epithelial cells, including the proximal convoluted tubule, loop of Henle, and distal convoluted tubule, during renal injury by increasing mitochondrial biogenesis in epithelial cells. Bowman’s capsule, podocytes, and endothelial cells have a narrow PGC-1α tolerance. Increased PGC-1α levels in podocytes induce podocyte proliferation and collapsing glomerulopathy development, whereas increased PGC1-α in endothelial cells alters endothelial function and causes microangiopathy, thereby resulting in renal injury. (B) The role of mitochondrial fusion and fission in mitophagy. Mitochondrial fusion is promoted by the Mfn2 gene, whereas Drp1 promotes mitochondrial fission. Increased Drp1 and decreased Mfn2 expression exacerbates tubular damage, thereby contributing to kidney disease; however, studies have shown opposite results and inconsistencies. Whether PGC-1α transcriptionally regulates Drp1 and Mfn2 requires further research.
Figure 2.
Schematic description of PGC-1α function in renal homeostasis. (A) PGC-1α associated with nuclear respiratory factors 1 and 2 (NRF-1/2) has a protective role in renal epithelial cells, including the proximal convoluted tubule, loop of Henle, and distal convoluted tubule, during renal injury by increasing mitochondrial biogenesis in epithelial cells. Bowman’s capsule, podocytes, and endothelial cells have a narrow PGC-1α tolerance. Increased PGC-1α levels in podocytes induce podocyte proliferation and collapsing glomerulopathy development, whereas increased PGC1-α in endothelial cells alters endothelial function and causes microangiopathy, thereby resulting in renal injury. (B) The role of mitochondrial fusion and fission in mitophagy. Mitochondrial fusion is promoted by the Mfn2 gene, whereas Drp1 promotes mitochondrial fission. Increased Drp1 and decreased Mfn2 expression exacerbates tubular damage, thereby contributing to kidney disease; however, studies have shown opposite results and inconsistencies. Whether PGC-1α transcriptionally regulates Drp1 and Mfn2 requires further research.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113447
AMA Style
Cheng C-F, Ku H-C, Lin H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. International Journal of Molecular Sciences. 2018; 19(11):3447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113447
Chicago/Turabian StyleCheng, Ching-Feng, Hui-Chen Ku, and Heng Lin. 2018. "PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation" International Journal of Molecular Sciences 19, no. 11: 3447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113447
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.