The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions


1
Department of Biochemistry & Molecular Biology and Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan 33302, Taiwan
2
Liver Research Center, Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan
3
Division of Allergy, Immunology and Rheumatology, Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan
Int. J. Mol. Sci. 2018, 19(12), 3940; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123940
Submission received: 13 November 2018
/
Revised: 5 December 2018
/
Accepted: 5 December 2018
/
Published: 7 December 2018
(This article belongs to the Special Issue Hepatitis Virus Infection and Molecular Research 2018)
Abstract
:Autophagy is an evolutionarily conserved cellular process in which intracellular components are eliminated via lysosomal degradation to supply nutrients for organelle biogenesis and metabolic homeostasis. Flavivirus infections underlie multiple human diseases and thus exert an immense burden on public health worldwide. Mounting evidence indicates that host autophagy is subverted to modulate the life cycles of flaviviruses, such as hepatitis C virus, dengue virus, Japanese encephalitis virus, West Nile virus and Zika virus. The diverse interplay between autophagy and flavivirus infection not only regulates viral growth in host cells but also counteracts host stress responses induced by viral infection. In this review, we summarize the current knowledge on the role of autophagy in the flavivirus life cycle. We also discuss the impacts of virus-induced autophagy on the pathogeneses of flavivirus-associated diseases and the potential use of autophagy as a therapeutic target for curing flavivirus infections and related human diseases.
1. Introduction
Autophagy is a lysosome-mediated catabolic process in which unwanted intracellular components are degraded to recycle nutrients for the regeneration of organelles and energy [1,2]. A variety of stresses, including nutrient starvation, organelle damage, accumulation of unfolded proteins and pathogen infection, have been demonstrated to activate autophagy [3,4]. Dysregulation of autophagy has been shown to play a role in the pathogeneses of numerous human diseases, such as cancer, neurodegenerative diseases and pathogen infection [5,6]. An increasing number of studies indicate that microbial infection activates autophagy, which degrades the invading microorganism and induces the innate immune response, thus restricting pathogen infection [7,8,9,10]. On the other hand, autophagy has emerged as a pro-viral pathway by which viruses activate to benefit viral growth in infected cells [11,12,13]. Approximately seventy enveloped, positive-strand RNA viruses belonging to the Flaviviridae family, including hepatitis C virus (HCV), dengue virus (DENV), Japanese encephalitis virus (JEV), West Nile virus (WNV) and Zika virus (ZIKV), that contain important human pathogens and thus exert a global burden on public health [14,15]. Flavivirus infections often induce rearrangement of the host cellular membrane to establish a membranous structure for viral growth, thus triggering a variety of stress responses in infected cells [16,17]. In recent years, emerging lines of evidence have shown that autophagy is activated by flavivirus infections to promote viral replication and counteract virus-induced stresses, such as protein unfolding and organelle damage [16,17]. Therefore, comprehensively understanding the interplay between host autophagy and flavivirus infection will provide key information for curing viral pathogenesis. In this review, we summarize the current knowledge on how Flaviviridae viruses manipulate and subvert host autophagy in infected cells and address the functional role of autophagy in flavivirus-host interactions as well as in the pathogeneses of virus-associated human diseases. Finally, we discuss the therapeutic potential of autophagy modulation in the intervention of flavivirus infection.
2. Autophagy
Autophagy is referred to as the “self-digestion” process in which eukaryotic cells sequester intracellular materials within double-membraned vesicle-like structures and then deliver these materials to lysosomes for degradation. At least three types of autophagy have been identified, including macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) (Figure 1). Macroautophagy (hereafter referred to as autophagy) involves the delivery of cytoplasmic components to lysosomes for degradation via autophagic vacuoles [2,18]. Microautophagy involves the direct transport of cytosolic content to lysosomes via the invagination and scission of lysosomal membranes into the lumen [19,20]. CMA is a selective elimination process in which substrates containing the “Lys/Phe/Glu/Arg/Gln” (KFERQ) motif are recognized by a chaperone protein, heat shock cognate protein of 70 kDa (Hsc70) and delivered to the lysosomal lumen via the lysosomal membrane protein 2A (LAMP2A) [21,22]. Autophagy was discovered in the mid-1950s by Christine de Duve, the 1974 Nobel Laureate in Physiology or Medicine and coined in the early 1960s at the Ciba Symposium on Lysosome [23,24]. In addition, ultrastructural transmission electron microscopy (TEM) analyses demonstrated the morphogenesis of dense bodies with sizes similar to that of mitochondria in various types of tissues of animals [25,26,27,28]. These dense bodies morphologically represent double-membranous vesicle structures in which mitochondria and the endoplasmic reticulum (ER) are engulfed [26,28,29]. Shortly thereafter in the late 1970s, several groups showed that deprivation of amino acids and growth factors can induce autophagy [30,31]. Biochemical studies performed in the 1970s–1990s demonstrated that autophagy promotes the degradation of long-lived proteins, which correlates with a decreased supply of amino acids [32,33]. Meanwhile, several groups characterized the intracellular signaling processes and molecules associated with autophagy and identified 3-methyladenine (3-MA) as an autophagy inhibitor [34,35,36,37,38,39,40,41,42]. Moreover, the concept of a phagophore developed for double-membranous autophagic vacuoles was first described [43,44,45]. Yoshinori Ohsumi initiated the comprehensive identification of genes functioning in autophagy in his work to characterize autophagic vacuoles in Saccharomyces cerevisiae and genetically screen temperature-sensitive mutants that are defective in the formation of autophagic vacuoles in yeast cells [46,47,48]; Yoshinori Ohsumi was awarded the 2016 Nobel Laureate in Physiology or Medicine for his work. Approximately fifteen autophagy-related genes were identified as being required for the completion of autophagy in yeasts [47]. At the same time, several groups also uncovered autophagy-related genes (ATGs) functioning in humans and other eukaryotes and the nomenclature for ATGs among different eukaryotic species was unified [49,50,51,52,53]. Approximately forty ATGs have currently been identified, most of which are highly conserved in nearly all eukaryotes [53,54,55].
2.1. The Biogenesis of Autophagic Vacuoles
The stepwise process of vacuole biogenesis in autophagy initiates with an intracellular membrane rearrangement promoting the emergence of an isolation membrane (IM)/phagophore (Figure 1) [56,57,58]. The membranous structure of the IM/phagophore originates from many types of organelles, including the ER [59,60], mitochondria [61], Golgi apparatus [62], plasma membrane [63], recycling endosome [64,65] and mitochondria-associated ER membrane [66]. The newly formed phagophore subsequently elongates and encloses into a double-membranous vacuole, termed autophagosome (Figure 1) [67,68,69,70]. Then, the autophagosome fuses with lysosomes (Figure 1), generating mature autolysosomes that degrade the sequestrated cargo by acidic proteases [69,71,72,73]. The entire autophagic process relies on the coordinated actions of ATGs to rearrange membranes for vacuole biogenesis as well as on multiple cell signaling pathways [18,74,75]. When cells are starved of nutrients, autophagy is activated by the repression of mammalian target of rapamycin (mTOR), a serine/threonine protein kinase required for metabolic regulation. The suppression of mTOR induces translocation of the unc-51 like-kinase (ULK) complex, which is organized by ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101, from the cytosol to ER membrane-reconstituted compartments (Figure 1) [76,77]. Subsequently, the class III phosphatidylinositol-3-OH kinase (PI3K) complex (class III-PI3K, including Vps34/PI3KC3, Vps15, Beclin 1 and ATG14) is recruited to the ER-derived nucleation site, triggering the generation of phosphatidylinositol-3-phosphate (PtdIns(3)P) (Figure 1) [75,78,79]. The newly formed PtdIns(3)P, in turn, recruits the double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI, the mammalian orthologue of ATG18) family proteins to generate an ER-associated omegasome structure, also known as the IM/phagophore (Figure 1) [80,81]. Elongation and enclosure of the IM/phagophore into a mature autophagosome relies on two ubiquitin-like (UBL) conjugation systems (Figure 1) [82,83,84,85]. ATG7 (E1) and ATG10 (E2) confer the conjugation of ATG12-ATG5, which interacts with ATG16L to form the ATG12-ATG5-ATG16L trimeric complex (Figure 1) [82,86]. At the same time, the phosphatidylethanolamine (PE)-conjugation of ATG8 family proteins (including the microtubule-associated protein 1 light chain 3 (LC3) and gamma-aminobutyric acid receptor-associated protein (GABARAP) subfamilies) initiates with the proteolytic cleavage of their C-termini by ATG4 family proteins [87,88]. Then, the cleaved ATG8 family proteins are conjugated to PE via the catalytic cascade of ATG7 (E1) and ATG3 (E2), producing the lipidated forms of ATG8 family proteins [87,88]. Finally, autophagosomes fuse with lysosomes to form mature autolysosomes (Figure 1), in which the sequestrated materials are degraded and recycled for nutrients.
The fusion of autophagosomes with lysosomes is also coordinated by multiple protein-protein interactions, cytoskeleton-mediated transport and membrane rearrangement events [70,71,73] (Figure 1). This fusion step initiates with the timely transport of autophagosomes and lysosomes via microtubules [89,90]. The small GTPase Ras-related protein 7 (Rab7) located on the surface of the autophagosome bridges the movements of the autophagosome on microtubules by binding to FYVE and coiled-coil domain-containing 1 (FYCO1) and Rab-interacting lysosomal protein (RILP) [91], which are respectively linked to kinesin and dynein on the microtubules [92,93,94,95]. Moreover, Rab7 located on late endosomes and lysosomes could be recruited to mature autophagosomes, thus promoting autophagosome-lysosome fusion [92,93]. The PI3K protein complex associated with UV radiation resistance-associated (UVRAG) also participates in the fusion of autophagosomes and lysosomes by interacting with Vps16, a subunit of the homotypic fusion and protein sorting (HOPS) complex [79,96,97]. In contrast, the binding of Rubicon to the PI3K protein complex inhibits autophagosome-lysosome fusion [79]. A further study highlights that ATG14L binds to syntaxin 17 (STX17) and synaptosome-associated protein 29 (SNAP29) binary complexes on the surface of autophagosomes, promoting STX17/SNAP29/vesicle-associated membrane protein 8 (VAMP8)-mediated autophagosome-lysosome fusion [98]. Furthermore, the pleckstrin homology domain-containing protein family member 1 (PLEKHM1) protein that contains an LC3-interacting motif has been shown to concomitantly interact with Rab7/HOPS and LC3, facilitating the fusion of autophagosomes and lysosomes [99]. Although the functional ATGs and other cellular proteins involved in the autophagic process have been extensively studied and characterized, the detailed regulatory mechanism underlying the functional roles of ATGs at each step of autophagy requires further investigation.
2.2. Selective Autophagy
In addition to bulk and nonselective degradation, increasing evidence indicates that autophagy can selectively eliminate specific cargos, including organelles and proteins [100,101,102]. Selective autophagy initiates with the recognition of cargo that are highly polyubiquitinated via cargo receptors and delivers degraded cargos to autophagic machinery via the binding of cargo receptors to ATG8 family proteins located on the autophagosomal membrane of IM/phagophore (Figure 2) [91,103,104,105]. Several cargo receptors of selective autophagy, such as neighbor of BRCA1 (NBR1), calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2, also known as NDP52), p62/sequestosome 1 (SQSTM1) and optineurin (OPTN), contain the LC3-interacting regions (LIR) required for interaction with ATG8 family proteins, thus promoting the engulfment of cargos into autophagosomes [91,105,106]. Despite this LIR-containing cargo receptor-mediated clearance of ubiquitinated substrates, several potential ATG8-interacting motifs (AIMs) and GABARAP-interacting motifs (GIMs) have been identified within ATGs and other cellular proteins [107,108,109,110]. Recently, the AIMs located within Saccharomyces cerevisiae ATG19 were shown to directly bind to ATG5, thus recruiting the ATG5-ATG12-ATG16L complex to cargo and stimulating the PE conjugation of ATG8 for closure of the autophagosome [111]. Whether LIRs, AIMs and GIMs participate in the completion of autophagy and recruitment of potential substrates to the selective autophagy process requires further study.
Selective autophagy plays a pivotal role in maintaining the integrity of intracellular organelles by degrading damaged organelles (Figure 2) [100,102,112]. The selective elimination of organelles, termed organellophagy, supplies recycled nutrients for the regeneration of mitochondria, peroxisomes, ER, lipid droplets (LDs), ribosomes, lysosomes and nuclei (Figure 2). The removal of mitochondria by selective autophagy, termed mitophagy, could be activated by hypoxia [113,114], accumulation of reactive oxygen species (ROS) [115,116,117] and mitochondrial depolarization [118,119,120]. The major route for clearing damaged mitochondria originates from the loss of PTEN-induced putative kinase 1 (PINK1) cleavage by presenilin-associated rhomboid-like protein (PARL) within the inner mitochondrial membrane and the inhibition of PTEN degradation via the ubiquitin-proteasome pathway [121,122]. The PINK1 accumulated on the outer mitochondrial membrane, in turn, phosphorylates ubiquitin and then recruits the ubiquitin E3 ligase Parkin [118,119,120,123,124,125]. Parkin, in turn, ubiquitinates the surface proteins on the outer mitochondrial membrane [118,119,120,123,126], triggering the recognition of cargo receptors for the removal of mitochondria by autophagy [127]. PINK1 specifically recruits Calcoco2/NDP52 and OPTN to mitochondria and subsequently induces the translocation of phagophore-generating factors, including DFCP1 and WIPI, for autophagosome maturation proximal to degradative mitochondria [127]. Notably, the TANK binding kinase 1 (TBK1)-mediated phosphorylation of p62/SQSTM1 at serine residue 403 and OPTN at serine residues 177, 473 and 513 are critical for promoting mitophagy [128,129,130]. In addition to the PINK1/Parkin-mediated mitophagy pathway, several outer mitochondrial membrane proteins, such as FUN14 domain-containing 1 (FUNDC1), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L) and yeast ATG32, have been shown to mediate mitophagy in a ubiquitin-independent manner [131,132,133,134,135]. Not surprisingly, several new molecules were recently identified as new cargo receptors of mitophagy, including prohibitin 2 (PHB2) and Toll-interacting protein (Tollip) [136,137]. Reciprocally, the dequbiquitination enzymes USP30 and USP35 have been shown to antagonize the mitophagy process by deubiquitinating Parkin [138,139].
In addition to mitophagy, several types of organelles have been shown to be removed by selective autophagy (Figure 2). The cognate cargo receptors responsible for the clearance of these organelles have been recently identified. For pexophagy, known as the degradation of peroxisomes by selective autophagy, yeast ATG36 and mammalian NBR1 and p62/SQSTM1 function as adaptors for the recruitment of damaged peroxisomes to autophagic degradation machinery (Figure 2) [140,141,142,143]. Multiple types of kinases, such as yeast Hrr25 and mammalian Ataxia-telangiectasia-mutated (ATM), phosphorylate these cargo receptors, facilitating their interaction with the autophagosome membrane [144,145]. Moreover, polyubiquitination of several peroxisomal (PEX) membrane proteins, such as PEX5 and the 70-kDa PEX membrane protein (PMP70), serves as a signal for recognition by cargo receptors [145,146]. The degradation of stressed ER by ER-phagy relies on the functions of yeast ATG39, ATG11 and ATG40 [147] as well as on the family with sequence similarity 134, member B (FAM134B) and reticulon family proteins (Figure 2) [148,149]. Notably, ATG39 and ATG11 have also been shown to participate in the selective degradation of yeast nuclei (Figure 2) [147]. Selective autophagy also involves the clearance of protein aggregates through p62/SQSTM1 and histone deacetylase 6 (HDAC6)-mediated recognition of Lys63 (K63)-linked poly-ubiquitin chain on aggregated proteins [150,151,152,153] (Figure 2). In addition, NBR1 and the autophagy-linked FYVE (ALFY) are shown to cooperate with p62/SQSTM1 to eliminate protein aggregates [154,155,156,157]. Lysophagy was recently identified as a new pathway for removing injured lysosomes via the concerted recruitment of galectin-3 and LC3 onto lysosomal membranes, as these proteins are presumably recognized by p62/SQSTM1 and targeted for degradation via autophagy (Figure 2) [158,159]. Very recently, autophagy has emerged as a critical pathway for the degradation of ribosomes, termed ribophagy (Figure 2) [160,161]. In addition, autophagy has been shown to catabolize LDs to maintain metabolic homeostasis (Figure 2) [162,163]. CMA also regulates the degradation of LDs via the 5′-AMP-activated protein kinase (AMPK)-mediated phosphorylation of perilipin 2, promoting recognition of the CMA chaperone Hsc70 for degradation [164]. Moreover, the newly identified cargo receptor of autophagy, nuclear receptor coactivator 4 (NCOA4) interacts with ATG8 family proteins and promotes degradation of the ferritin heavy and light chains, thus controlling intracellular ion levels (Figure 2) [165,166]. This autophagy-mediated turnover of ferritin is termed ferritinophagy, which has been suggested to regulate erythropoiesis and DNA replication in blood cells [167,168]. Elimination of invading microbes acts as the first line of host defense to counteract pathogen infection [169,170,171]. To this end, in the xenophagy process, invading pathogens are directly engulfed and eliminated via the p62/SQSTM1-, Calcoco2/NDP52- and OPTN-mediated recognition of ubiquitinated microbial proteins (Figure 2) [172,173,174]. This process also requires the phosphorylation of p62/SQSTM1 (at serine residues 349 and 403) and OPTN (at serine 177) [174,175,176,177]. Furthermore, autophagy also participates in the activation of an innate immune defense response to inhibit microbial infection, as it induces Toll-like receptor (TLR)-mediated immune responses and facilitates the presentation of antigens derived from pathogens to major histocompatibility complex (MHC) class II molecules [178,179,180,181,182]. Collectively, autophagy not only regulates cellular homeostasis via selectively removing harmful organelles from cells but also serves as a host defensive mechanism to restrict pathogen infection.
2.3. Autophagy and Diseases
Autophagy is a catabolic pathway utilized by eukaryotic cells to counteract stress responses, promote organelle turnover and eliminate aggregated proteins and excess lipids [1,2]. Moreover, autophagy also degrades infectious microbes and participates in regulating the immune response [100,102,112,183]. Therefore, autophagy protects cells from damage and maintains cellular homeostasis [5,6,183]. Dysregulation of autophagy has been implicated in the development of various types of human diseases, including cancer [183,184], neurodegenerative diseases [185,186,187,188], infectious diseases [7,172], cardiovascular diseases [189,190,191], aging [184,185,189,190,191,192,193] and metabolic disorders [183,194,195,196,197]. Thus, the modulation of autophagy presents a valuable and potential therapeutic target for treating human diseases. However, multiple obstacles, including drug specificity, the limited in vivo models for testing drug potency and unexpected side effects, hamper the implementation of autophagy activators and inhibitors in the clinic. Comprehensively understanding the entire autophagic process will provide more information for the development of rationally designed autophagic modulators in the future.
3. Flaviviridae Viruses
The Flaviviridae family comprises enveloped single-stranded, positive-sense RNA viruses that are approximately 50~70 nm in diameter [14,15,198] and includes four major genera: Flavivirus, Hepacivirus, Pestivirus and Pegivirus [15,198,199]. Several members of the Flavivirus and Hepacivirus genera are human pathogens that cause millions of infections annually and thus exert a global burden on public health [14,15,198]. HCV, a member of the Hepacivirus genus, is a leading cause of chronic hepatitis and at least 3% of the human population is infected by HCV worldwide [200,201]. Long-term chronic HCV infection often leads to multiple liver diseases, including steatosis, cirrhosis and hepatocellular carcinoma (HCC) [202,203]. The four prevalent human pathogens DENV, WNV, JEV and ZIKV belong to the genus Flavivirus [199,204,205]. These mosquito-transmitted viruses account for most severe arbovirus infections in the human population and irregular outbreaks exert an enormous threat on global health [205,206,207,208,209,210,211,212]. Primary DENV infection often causes reactions ranging from asymptomatic illness to symptoms such as rush, headache and dengue fever [205,206,213]. Secondary, homo- or heterotypic infections may develop into severe dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), most likely due to antibody-dependent enhancement (ADE), which increases viral infectivity and pathogenicity [205,206,213,214]. JEV is a major cause of encephalitis flavivirus in the Asia-Pacific region [207,208,215]. No significant symptoms are observed in the vast majority of JEV-infected individuals but severe clinical illness still occurs in rare cases of infection, resulting in an approximately 30% mortality rate, particularly in children [207,208,215]. Most remnant-infected patients who survive often suffer permanent neurological deficits, including seizures, paralysis and mental retardation [207,208,215]. WNV is an emerging and mosquito-transmitted flavivirus that causes fever and neuroinvasive diseases, including encephalitis and poliomyelitis [210,216] and serious illness in aged and immunocompromised patients often introduces a higher risk of mortality [217,218]. The recently re-emerged ZIKV led to large outbreaks in South and Central America from 2015–2016 [211,219]. The virulence of this re-emerged ZIKV strikingly increases the incidence rates of microcephaly in newborn infants and Guillain-Barré syndrome in adults and this virus thus serves as another threat to public health worldwide [220,221,222]. No approved vaccination strategies against these human pathogenic flaviviruses are available except for JEV [223,224]. Recently, oral direct-acting antiviral (DAA) drugs targeting HCV replication have been approved and shown to cure HCV infection in more than 90% of patients [225,226]. However, antiviral drugs for the clinical treatment of other flavivirus infections are still limited [227].
3.1. Hepatitis C Virus (HCV)
HCV was first discovered to be the infectious agent underlying non-A, non-B hepatitis in 1975 [228] and the HCV viral RNA sequence was cloned in 1989 [229]. Currently, seven isolates of HCV have been cloned and classified, including genotypes 1 through 7 and each genotype is further divided into an array of different subtypes [230]. The high genomic variability among the different HCV genotypes may lead to poor cross-genotype immunity and varying levels of disease progression [231,232]. In recent decades, the combined therapy of pegylated-interferon (IFN) and ribavirin has been standardized for treating HCV infection [233]. However, the genotype of the infecting virus, genetic polymorphisms, disease stage and severe side effects often hamper the successful rate of treatment [225,233,234]. IFN-free and DAA-based anti-HCV therapy has led to an HCV infection cure rate of more than 90% [225,226]. However, the emergence of resistance to DAA drugs [225,235] and the uncertain and controversial effects of DAA treatment on disease progression in chronically infected patients still impede the complete eradication of HCV infection [236,237]. For instance, data on the impact of DAA treatment on the risk for HCC occurrence in chronic HCV patients are still conflicting. Several studies have demonstrated that the rate of HCC development is significantly reduced in patients who have achieved a sustained virological response (SVR) after DAA therapy [238,239,240]. However, numerous reports have indicated that the rates of HCC recurrence and de novo development of HCC in patients after DAA-induced SVRR are unexpectedly increased [241,242], possibly due to uncontrolled liver immunity resulting from the DAA-mediated eradication of HCV-specific T cells [243].
The HCV viral genome comprises 9.6 Kb of positive-sense single-stranded RNA (ssRNA) that contains an open reading frame (ORF) and two untranslated regions (UTRs) located on the 5′- and 3′-termini [201,244,245,246]. The positive strand of the HCV RNA genome can be directly translated into a polypeptide of approximately 3300 amino acids and then processed into three structural proteins (core and envelope glycoproteins E1 and E2) and seven nonstructural (NS) proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) by a combination of cellular and viral proteases [201,244,246]. The core, E1 and E2 proteins are the major constituents responsible for assembly of the HCV virion [7,196,244,245], while the NS proteins are required for organization of the replication complex and reconstitution of membranous compartments, that is, a membranous web for HCV viral RNA replication [27,101,247,248]. Moreover, the concerted localization of NS proteins on the surface of LDs also participates in HCV viral particle assembly [249,250].
The entry of HCV into hepatocytes relies on several cell surface molecules, termed entry (co)receptors, including tetraspanin CD81 [251,252,253], scavenger receptor class B member 1 (SCRAB1) [254,255], tight junction proteins, claudin 1 (CLDN1) [256] and occludin (OCLN) [257]. Moreover, the lipoprotein-binding proteins, proteoglycan and leptin-binding protein located on the surface of hepatocytes, such as low-density lipoprotein receptor (LDLR) [258,259], heparan sulfate (HS) [260,261] and dendritic cell-specific intercellular adhesion molecule three grabbing nonantigen (DC-SIGN) [262], have been shown to facilitate the attachment of lipoprotein-associated HCV virions to the cell surface. Following the initial attachment to the cell surface, viral particles bind to SCARB1 and CD81 via interactions with the E1 and E2 structural proteins [263,264,265]. Then, the epidermal growth factor receptor (EGFR) and downstream effector GTPase HRas trigger the association of CD81 with CLDN1 to facilitate the binding of HCV to OCLN on the basolateral surfaces of hepatocytes [266,267,268,269,270]. The viral particle is subsequently internalized via a clathrin-mediated and pH-dependent endocytosis pathway [271,272]. After the viral envelope fuses with the endosomal membrane, HCV undergoes an uncoating process to release the viral genome into the cytoplasm, wherein viral RNA is translated and replicated [271,272]. Moreover, several additional molecules, such as ephrin receptor A2 (EphA2) [270], Niemann-Pick C1-like L1 (NPC1L1) cholesterol uptake receptor [273], transferrin receptor (TfR) [274], tetraspanin CD63 [275] and cell death-inducing DFFA-like effector B (CIDEB) [276], as well as two additional tight junction proteins, CLDN6 and CLDN9, could mediate the entry of HCV into peripheral blood mononuclear cells lacking CLDN1 expression [277,278]. Notably, the second extracellular loop of OCLN together with CD81 and CIDEB respectively contribute to the host and tissue tropism of HCV entry [257,276]. The detailed molecular mechanism by which these newly identified entry (co)receptors mediate HCV entry remains to be elucidated.
The development of HCV-related liver diseases is a complicated, long-term process that has not been completely elucidated. Primary HCV infection is often asymptomatic and self-limiting but severe acute hepatitis and fulminant hepatic failure do occur rarely [279,280,281,282]. Most HCV infections are persistent and 20~30% of chronically infected patients develop liver steatosis, fibrosis and cirrhosis [279,280,281,282]. Ultimately, 3~5% of infected individuals develop HCC [279,280,281,282,283,284,285,286]. In addition to liver-associated diseases, HCV chronic infection also leads to extrahepatic diseases, such as mixed cryoglobulinemia vasculitis [282,287,288] and is perhaps linked to the progression of metabolic disorders, that is, diabetes and insulin resistance [289,290].
3.2. Dengue Virus (DENV)
DENV, the major mosquito-borne human pathogen, infects approximately 3.6 billion people in more than 100 countries worldwide and is thus a public health problem [206,291,292,293]. Since DENV was first isolated by Ren Kimura and Susumu Hotta in 1943 [293,294], four major serotypes of DENV (DENV1-4) have been identified and their genomic sequences are at least 65% similar [295,296]. Transmission of DENV to vertebrate host cells relies on its natural hosts, the mosquito vectors Aedes aegypti and Aedes albopictus [275,276,296,297]. Most DENV primary infections are asymptomatic and self-limiting [187,188]. However, clinical symptoms ranging from subclinical infection to dengue fever (DF) to the most severe forms, severe DHF and DSS, threaten the lives of some DENV-infected patients [298,299,300]. DF is a predominant symptom after primary and secondary infections, accompanied by febrile seizures, headache, rash and retro-orbital pain lasting for 1~2 weeks [298,299,300]. DHF, a severe dengue symptom that often results from homologous reinfection or secondary infection by a different DENV serotype, is characterized by thrombocytopenia, liver injury and hemorrhagic manifestations that ultimately induce DSS, which may cause mortality [298,299,300]. To date, effective antiviral drugs that cure DENV infection and vaccinations against DENV infection are still unavailable and under developed [227,300,301,302].
The DENV virion, a spherical particle that is appropriately 50 nm in diameter, is enclosed by a lipoprotein envelope [295,297]. The DENV viral genome is approximately 11 kb in length and contains an ORF that encodes three structural proteins (capsid, pre-membrane (prM) and glycosylated envelope E) and seven NS proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [291,297]. Particularly, the E protein harbors the membrane fusion ability to help virions attach to the host cell surface, interact with entry factors and assemble viral particles [303,304]. DENV infection of the host cell initiates with E protein-mediated receptor binding followed by clathrin-mediated endocytosis [305,306]. The diverse molecules present on the cell surface, including HS [307], DC-SIGN [308,309], mannose receptor (MR) [310], the lipopolysaccharide (LPS) receptor CD14 [311], the heat shock proteins 70 (HSP70) and 90 (HSP90) [312], 78-kDa glucose-regulated protein (Grp78) [313], lectin [314,315,316], laminin receptor [317], T cell immunoglobulin domain and mucin domain (TIM) and Tyro3, Axl and Mertk (TAM) phosphatidylserine receptors [318,319], as well as the tight junction protein CLDN1 [320,321] mediate the attachment of DENV and facilitate the entry of the virus into cells. The broad expression panel of these entry-associated factors in different types of tissues implies the low specificity for DENV entry receptors and the wide range of DENV tissue tropism.
After entering host cells, the viral genome is then released from the late endosome into the cytosol [305,306] and used as a template for translation and viral replication. Notably, the nondegradative ubiquitination of the DENV capsid protein is required for viral genome uncoating [322]. The NS proteins involves in the replication of viral RNA and the structural proteins assemble with the nascent viral RNA to generate infectious particles [15,300,323,324]. In a similar fashion to HCV, the DENV capsid protein also destabilizes the ER-derived membranous structure and LDs for virion assembly [325,326,327]. Due to the lack of an antiviral drug with high potency and an available vaccine for effective intervention against DENV infection, more efforts focused on understanding the DENV-host interactions will promote the exploration of potential therapeutic targets and new drug development [227,300,301,302]. In addition, elucidation of DENV-induced host cellular responses is urgently needed to delineate the pathogenesis of DENV-associated diseases.
3.3. Japanese Encephalitis Virus (JEV)
JEV is an infectious agent that remarkably leads to severe neurological disorders, affecting individuals in approximately 25 Asian countries [212,215]. JEV infection is considered asymptomatic in most infected individuals but a spectrum of clinical symptoms ranging from acute febrile seizure onset to severe encephalitis can appear [207,208,215]. Acute encephalitis often causes persistent neurological damage and mental disorders, such as parkinsonian syndromes and flaccid paralysis [207,208,215]. Based on nucleotide sequence similarities in the viral genome, five genotypes of JEV have been identified and divided (I through V) and infections by each genotype are prevalent in different regions [328]. JEV, first discovered and isolated in 1935 [329], is enzootically transmitted among mosquitoes (the genus Culex) and vertebrate hosts, including wild wading birds, cattle and pig [212,215,328]. JEV is a small enveloped flavivirus that is 50 nm in diameter. The JEV genome comprises approximately 11 Kb of positive-sense ssRNA that contains an ORF flanked by 5′- and 3′-UTRs [212,215]. After virus entry into permissive cells, the viral RNA can be translated into an ~3400 amino acid polypeptide precursor, which is co- and post-translationally processed by host signal peptidases and viral proteases into three structural proteins (capsid, prM and envelope E) and seven NS proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [212,215,330,331]. JEV shares a viral entry feature common to most flaviviruses, as it relies on virion attachment and interactions of the E protein with entry factors on the host cell surface [332,333]. In a similar fashion to DENV infection, the entry of JEV into a wide spectrum of susceptible cells requires several host molecules, including glucoaminoglycans (GAGs) [334,335,336], C-Type lectins [337,338,339,340], integrins [341], HSP70 [342,343,344], Grp78 [345], CD14 [346] and the intermediate filament vimentin [347,348]. Several types of vaccination strategies have been used for the clinical intervention of JEV infection [215,349]. However, virus replication-targeted antiviral treatment other than the currently supportive care for JEV-induced encephalitis is still limited and urgently needs further development.
3.4. West Nile Virus (WNV)
WNV is a neurotropic, arthropod-borne flavivirus that is transmitted in an enzootic cycle between mosquitoes, birds and other vertebrates [210,350]. WNV was first discovered and isolated from a female patient in the West Nile district of Uganda in 1937 [351]. WNV is also a human pathogen that may lead to significant morbidity and mortality [210,350,352]. The WNV viral genome comprises positive-sense ssRNA that is ~11 Kb in length [210,350]. WNV genomic RNA is used as a template for the translation of a polypeptide, which is subsequently processed by a combination of cellular and viral proteases to generate three structural proteins (capsid, prM and envelope E) and seven NS proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [210,350]. Analogous to other members of the Flavivirus genus, the structural proteins confer the encapsidation of viral RNA to form a virion, whereas NS proteins participate in replication of the viral genome [210,350,352]. WNV infection of host cells depends on several entry factors, such as GAGs [353], DC-SIGN [354,355], C-type leptin [356] and integrin [357,358]. In humans, WNV infection is often asymptomatic but approximately 20% of infected individuals suffer symptoms that range from febrile seizures to severe neurological disorders [210,350,352,359]. In rare cases of WNV infection, severe neurological manifestations can lead to death [210,350,352,359]. Thus far, a vaccine for WNV intervention and clinically approved drugs for eradicating WNV infection remain in development.
3.5. Zika Virus (ZIKV)
ZIKV is a mosquito-borne human pathogen that was first isolated from humans in 1954 [360]. ZIKV is an enveloped virus that harbors a positive-sense ssRNA that is ~10 Kb in length [211,361,362]. The viral RNA genome contains UTRs at the 5′- and 3′-termini and an ORF [14,15,198,209,221]. The viral RNA can be used as a template for translation of a polypeptide that is further cleaved by host and viral proteases into three structural proteins (capsid, prM and envelope E) and seven NS proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [162,167,195,220,261]. The structural proteins participate in assembly of the virion, while the NS proteins are responsible for viral RNA replication, viral-encoded nascent polypeptide processing and modulating the host cellular response [123,187,304,317,354]. Because the amino acid sequences of the DENV E and ZIKV E proteins are more than 50% similar, ZIKV infection may use a similar route and entry factors, such as TAM phosphatidylserine receptor [363,364] and GAGs [365], function in the entry of DENV into host cells [332,366]. ZIKV infection in humans is considered an asymptomatic illness but 10~20% of infected patients develop flu-like symptoms 7~14 days after infection [14,15,198,209,221]. However, this rate was enhanced by nearly 50% among individuals infected by the ZIKV that re-emerged in America from 2013~2015, characterized by febrile seizures, organ damage, encephalitis and thrombocytopenia [14,15,198,209,221,367]. Most importantly, ZIKV infection in pregnant patients leads to microcephaly and congenital ZIKV syndrome in newborn infants [14,15,198,209,221,367]. Notably, ZIKV infection in adults results in Guillain-Barré syndrome [368,369,370,371,372,373,374,375]. Antiviral drugs for curing ZIKV are still unavailable but multiple kinds of vaccines have been initially developed and are anticipated to combat ZIKV infection [376,377,378,379,380].
4. Flavivirus-Autophagy Interactions
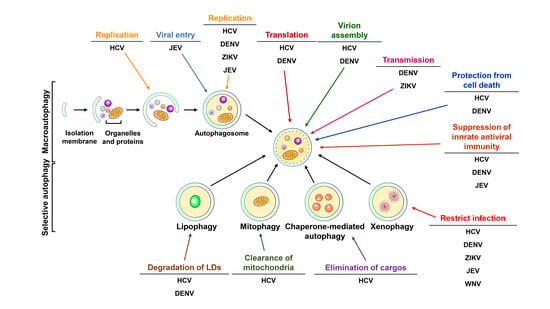
The virus-induced modulation of autophagy was first discovered in the mid-1960s [381]. Ultrastructural electron microscopy analysis revealed immense membraned-enclosed bodies in poliovirus-infected HeLa cells [381]. The authors also showed that these virus-induced double-membraned bodies associate with virions [381]. Soon after this study, these vesicle-like structures associated with lysosomal enzymes were extensively observed in picornavirus- and herpesvirus-infected tissues and cells [382,383,384,385,386]. During the 1990s–2000s, several studies demonstrated that poliovirus infection induces autophagic process to support viral-induced membranous structure formation [387,388]. Later, Levine and colleagues first identified that the herpes simplex virus-1 (HSV-1) neurovirulence protein ICP34.5 can antagonize elongation initiation factor alpha (eIF2 alpha) kinase (also known as PKR)-mediated autophagy activation [389]. Prentice et al. showed that coronavirus mouse hepatitis virus (MHV) induces autophagy to promote the formation of a replication complex for MHV growth [390]. Christian Münz’s group demonstrated that autophagy participates in the delivery of Epstein-Barr virus nuclear antigen 1 (EBNA1) to lysosomes, contributing to the processing of MHC class II molecules [179]. The suppressive effect of autophagy inhibition via 3-MA on viral replication was also demonstrated in poliovirus- and rhinovirus-infected cells [391]. Human parvovirus B19 was also shown to activate autophagy to protect infected cells from viral-induced cell death [392]. In contrast to its pro-viral function, autophagy was also shown to play an antiviral role by inhibiting virus infection in the mid-2000s. Talloczy et al. provided the first line of evidence that xenophagy can eliminate HSV-1 via the eIF2alpha-PKR signaling pathway [393]. In addition to supporting virus replication and degrading the infecting virus, autophagy also contributes to the apoptosis of CD4+ T lymphocytes after human immunodeficiency virus (HIV) envelope proteins bind to C-X-C chemokine receptor type 4 (CXCR-4) in infected cells [394], suggesting that virus infection can trigger the HIV-induced destruction of CD4+ T cells. On the other hand, autophagy is also thought to mediate the recognition of ssRNA viruses and the production of IFN by plasmacytoid dendritic cells (pDCs) via TLR7 [178]. These results collectively suggest that autophagy plays diverse roles in virus-host interactions by promoting viral replication via supporting membranous compartments, degrading infectious viruses via xenophagy, enhancing antiviral immunity by enhancing MHC-mediated antigen presentation, protecting infected cells from death and triggering viral infection induced by the elimination of T lymphocytes [9,11,13,395,396,397]. In the late 2000s, numerous studies demonstrated that autophagy is induced by Flaviviridae, among which HCV and DENV were the first shown to activate autophagy and thus benefit the viral life cycle [398,399,400]. To date, nearly one hundred studies have investigated the role of flavivirus-infected autophagy in the viral life cycle and host cell responses. These studies indicate that flavivirus-activated autophagy may participate in the replication of viral RNA, translation of viral RNA, the entry of virions and assembly of infectious particles. Moreover, autophagy also participates in modulation of the virus-induced antiviral immune response and elimination of organelles and proteins in flavivirus-infected cells. On the other hand, flavivirus infection also induces autophagy to trigger the xenophagy pathway and thus degrade infecting viruses. Although autophagy plays a key role in flavivirus-host interactions, controversy and discrepancies exist among studies. Thus, we comprehensively summarize the current knowledge of the interplay between flaviviruses and host autophagy and the functional impacts on virus replication and host response. We also discuss the molecular mechanism by which flaviviruses drive autophagy to promote their life cycle and the pathogeneses of flavivirus-related diseases.
4.1. HCV
In the past decade, numerous studies have extensively shown that HCV modulates autophagy to promote the viral life cycle and alter cellular signaling. Ait-Goughoulte and colleagues first showed that serially passaging HCV genotype 1a (clone H77) in immortalized human hepatocytes (IHH) induces autophagic vacuole formation, as shown by TEM analysis and enhanced the generation of GFP-LC3-labeled autophagosomes, as determined by immunofluorescence (IF) assays (Table 1) [399]. Not only was the number of autophagic cells increased but the levels of Beclin 1 and ATG12-ATG5 conjugate were also elevated in HCV/H77-replicated IHH cells [399]. Soon afterward, transfection of a full-length HCV genotype 2a JFH1 viral RNA into human hepatocellular carcinoma Huh7 cells was reported to trigger incomplete autophagy, as demonstrated by the increased formation of autophagosomes rather than autolysosomes, inhibition of autophagic flux and degradation of long-lived proteins (Table 1) [401]. In addition, the authors also demonstrated that the unfolded protein response (UPR) is required for HCV replication-induced incomplete autophagy [401]. Moreover, their study also indicated that individually knocking down UPR- and autophagy-related genes significantly repressed the replication of HCV viral RNA [401]. To understand whether HCV JFH1 infection indeed induces autophagy in Huh7 cells, Chisari’s groups first analyzed the impact of cell culture-derived infectious HCV (HCVcc) on host autophagy in Huh7.5.1 cells, a clone derived from Huh7 cells that is highly permissive to HCV replication [402,403,404]. They first demonstrated that HCVcc JFH1 infection can induce the autophagy required for translation of the incoming viral RNA to establish virus infection (Table 1) [405]. Moreover, no colocalization of HCV NS proteins with viral-induced GFP-LC3-labeled autophagic vacuoles was observed, suggesting that the HCV-induced autophagic membrane does not primarily provide the replication compartment for HCV replication [405]. Ke and Chen demonstrated that HCVcc JFH1 infection induces the entire autophagic process throughout the formation of mature autolysosomes [405,406]. This study showed that HCV infection enhances autophagosome and autolysosome formation and increases autophagic flux [405,406]. Moreover, interference with autophagy using gene silencing and pharmacological inhibitors strikingly inhibits HCV viral RNA replication rather than the translation of viral RNA [405,406]. Most importantly, the HCV pathogen-associated molecular pattern-mediated IFN response was elevated by repressing HCV-induced autophagy in infected cells, suggesting that HCV may induce complete autophagy to suppress innate antiviral immunity [405,406]. Consistent with this study, Shrivastava et al. reported that genetically silencing ATGs, such as Beclin and ATG7, reduces HCV infectivity and activates IFN-stimulated gene expression in HCV H77-infected IHH cells (Table 1) [407]. This conclusion was further strengthened by a recent study showing that HCV-induced autophagy degrades tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) via p62/SQSTM1, suppressing host innate immunity (Table 1) [408]. Collectively, these studies indicate that HCV-activated autophagy may suppress antiviral innate immunity to promote HCV replication [405,406,407,409].
Apart from repressing the antiviral response to support viral replication, several studies have suggested that HCV-induced autophagy may enhance viral RNA replication via interactions between autophagic machinery and viral proteins (Table 1) [411,412]. Guevin and colleagues found that ATG5 transiently interacts with NS5B and NS4B during the early stage of HCV infection and that silencing ATG5 downregulates the intracellular amount of viral RNA in infected cells, implying that autophagy promotes the initial replication of nascent viral RNA by affecting NS5B polymerase activity and the NS4B-altered membranous web, which are necessary for HCV replication [412]. Another study further showed that the HCV NS proteins-organized replication complex along with viral RNA localize within the HCV-induced autophagosome [411], suggesting that HCV-activated autophagy provides a resource for reconstituting the membrane compartment to replicate HCV viral RNA. In agreement with this study, Sir et al. showed that HCV NS5A, NS5B and HCV viral RNA colocalized with GFP-LC3-labeled autophagosomes by IF analysis [411]. These authors further demonstrated that HCV viral RNA localizes to the autophagosome membrane using coimmunoprecipitation and immunogold-TEM assays [411], in agreement with another study showing that HCV may activate autophagy to promote the biogenesis of double-membraned vesicles (DMVs) that support the replication of HCV viral RNA (Table 1) [413]. Very recently, a biochemical fraction study demonstrated that HCV infection triggers the translocation of lipid rafts to autophagosomes to promote viral replication [414]. In contrast, Bartenschlager′s research group demonstrated no significant colocalization between autophagic vacuoles and HCV NS proteins or the dsRNA replicative intermediate [435] and that the generation of HCV-induced DMVs are associated with ER and LDs rather than with autophagic vacuoles in HCVcc-infected and replicon cells [436]. These results coincide with those of other studies showing that HCV-triggered autophagic vacuoles do not colocalize with HCV replication complex components [405,437]. These studies imply that the viral-induced autophagic process is not required for organization of the HCV viral RNA replication platform.
Autophagy was also shown to promote HCV virion assembly. Tanida et al. showed that knockdown of ATG7 and Beclin 1 moderately reduces the extracellular infectivity of infected cells without affecting the intracellular expression of viral proteins and RNAs (Table 1) [415], implying that HCV-induced autophagy may help the egress and release of mature virions. This observation was further supported by Shrivastava’s study, which showed that HCV-activated autophagy participates in the budding of infectious viral particles via the CD63-associated exosome pathway (Table 1) [416]. Moreover, Kim and Ou showed that HCV induces autophagy to regulate apolipoprotein E (ApoE) transport and thus promote HCV virion assembly (Table 1) [417].
Furthermore, autophagy was shown to be differentially activated in a genotype-dependent manner in HCV replicon cells, supporting viral replication rather than the complete viral life cycle (Table 1) [418]. Additionally, the replication of HCV Con1 (genotype 1b) replicon RNA interferes with autophagy maturation and impedes the secretion of cathepsin upon autolysosome maturation [418]. Moreover, inhibiting the HCV Con1 replicon-induced autophagosome by ectopically expressing ATG4BC47A, a mutant ATG4B that inhibits the lipidation of ATG8 family proteins, results in severe cytoplasmic vacuolation and cell death [418], suggesting that HCV may utilize autophagy to counteract cell death [418]. Notably, this study mentioned the differential impacts of HCV viral RNA replication (Con 1b replicon) and infectious HCVccon autophagy [418].
Apart from the replication of viral RNA and infectious HCVcc, several HCV viral proteins have also been shown to induce autophagy. Su and colleagues reported that HCV NS4B is the only viral protein that sufficiently induces incomplete autophagy via residues 1~190 (Table 1) [419]. Gregoire et al. showed that autophagy can be activated by ectopic expression of HCV NS3 alone (Table 1) [420]. In addition to NS4B and NS3, expression of the HCV core protein can induce complete autophagy (Table 1) [421,422] and another later study showed that HCV NS5A can trigger autophagy (Table 1) [423,424]. Although these results collectively suggest that HCV can activate autophagy via individual viral proteins, many conclusions were drawn based on the transient expression of viral proteins in cells that are not permissive to complete HCV life cycle, which may have led to large discrepant results.
The molecular mechanism by which HCV exploits to initiate autophagy has been investigated. Several groups have reported that HCV may activate ER stress, which is required for autophagy activation (Table 1) [401,405,406,409]. In addition to this mechanism, UPR inhibitors have been shown to suppress HCV viral RNA replication and viral-induced autophagy [438]. Wang et al. further reported that the HCV core protein triggers the UPR to upregulate DNA damage-inducible transcript 3 protein (DDIT3, also known as CHOP) expression, which consequentially upregulates the transcription of LC3B and ATG5 to activate autophagy (Table 1) [421]. In contrast to these studies, Bjorn-Patrick and colleagues reported that HCV infection activates autophagy independent of the UPR and that the UPR is not required for HCV growth [437].
HCV was also shown to induce ER stress to interfere with protein kinase B (PKB)-tuberous sclerosis (TSC)-mTOR complex 1 (mTORC1) signaling and thus activate autophagy (Table 1) [439]. Analogously, Shrivastava and colleagues also showed that HCV induces autophagy by elevating Beclin 1 expression and activating mTOR signaling (Table 1) [440]. Apart from ATGs functioned in autophagosome biogenesis, their physiological role in phagophore formation and autolysosome maturation in HCV-autophagy interactions has been studied. STX17, which functions in the autophagosome-lysosome fusion process, was first shown to control HCV egress by regulating the equilibrium between the release of a mature virion and the degradation of intracellularly retained viral particles within the lysosome (Table 1) [425]. On the other hand, Wang and colleagues showed that the STX17-mediated homotypic fusion of phagophores enhances HCV-induced autophagosome formation to benefit HCV viral RNA replication (Table 1) [441]. Very recently, alcohol was shown to enhance PIAS family protein (PIASy) expression to activate autophagy and thus promote HCV replication (Table 1) [426]. Recent studies implied that alternatively spliced forms of ATG10 can differentially modulate autophagic flux to regulate HCV replication (Table 1) [427,428].
In addition to acting as a pro-viral factor benefiting viral growth, autophagy has also been shown to eliminate LDs and mitochondria (Table 1) [429,430]. Vescovo et al. first showed that the expression of an autophagy marker, the lipidated LC3, is oppositely correlated with the clinical parameters related to steatosis in liver biopsies of chronic HCV-infected patients (Table 1) [429]. Their study further demonstrated that autophagy promotes the degradation of LDs in HCV replicon cells [429], suggesting that HCV-activated autophagy catabolizes LDs to circumvent the HCV-induced excess of LD accumulation in infected cells [429]. In addition, HCV was also shown to enhance mitophagosome formation to eliminate mitochondria in infected cells in a PINK-Parkin-dependent manner (Table 1) [430,442]. Moreover, this PINK-Parkin-mediated clearance of mitochondria by autophagy is required for replication of HCV viral RNA in infected cells [430]. Further study demonstrated that HCV triggers mitochondrial fission to promote the Parkin-mediated degradation of mitochondria, thus attenuating cell apoptosis and establishing viral persistence (Table 1) [431]. In contrast, Hara et al. reported that the HCV core may interact with Parkin to interfere with the translocation of Parkin into mitochondria, thus alleviating mitophagy and sustaining HCV-triggered mitochondrial damage (Table 1) [432]. Very recently, the replication of HCV viral RNA was implicated to trigger selective autophagy to engulf ubiquitinated aggregates within the viral-induced autophagic vacuoles (Table 1) [443]. On the other hand, HCV-induced CMA was shown to participate in the degradation of IFN-alpha receptor-1 (IFNAR1), which is stimulated by free fatty acid (FFA) (Table 1) [433]. Moreover, Matsui and colleagues recently reported that HCV NS5A can interact with Hsc70, a regulator of CMA, to target hepatocyte nuclear factor 1α (HNF1α) for lysosomal degradation (Table 1) [424]. A recent study implied that HCV-induced IFN-β-inducible SCOTIN recruits NS5A to autolysosomes for degradation, thus restricting HCV replication (Table 1) [434]. Together, these studies imply that HCV may utilize autophagy to counteract active viral replication and the host stress response by eliminating specific cargos.
Little is known about whether HCV indeed activates autophagy to regulate virus-host interactions in vivo, although increasing evidence implies a major pro-viral role of autophagy in the HCV replication cycle in infected cells of the in vitro hepatocyte cell culture model. The lack of a reliable small animal system for investigating the HCV life cycle and host cellular response and the inconvenient monitoring of HCV-host interactions in HCV-infected liver specimens account for the slow progress in elucidating the detailed physiological role of autophagy in the HCV life cycle in HCV-infected cells. Notably, whether autophagy is activated in host cells infected by different HCV genotypes is still unresolved because the only infectious HCVcc used in the in vitro model, HCV JFH1 (genotype 2a), harbors an extraordinary replication cycle that is quite different from that of the prevalent genotypes 1 and 3 in most infected individuals [230,402].
In addition to promoting HCV growth, viral-triggered autophagy seems to degrade a panel of intracellular components, including LDs, mitochondria and host and viral proteins [424,429,430,434,443]. However, these conclusions are largely debated to the previous study shown that HCV activates incomplete autophagy by inhibiting autophagic flux [402,418]. Given that both HCV replication and autophagy require enormous rearrangement of the host intracellular membrane, whether these proteins and organelles are selectively engulfed into HCV-activated autophagic machinery before the specific cargo-degrading receptors are identified remains unknown. Moreover, whether the elimination of these cargos participates in the development of HCV-associated liver diseases also requires further elucidation.
Autophagy has emerged as a critical player in balancing lipid metabolism by the selective degradation of LDs, that is, lipophagy [163] and by regulating LD biogenesis [444,445]. Moreover, further study showed that autophagy controls the accumulation of bodily lipids and regulates adipocyte differentiation [446]. Together, these results indicate that alteration of autophagy may interfere with lipid catabolism and metabolic homeostasis. Furthermore, autophagy has been negatively correlated with the clinical parameters of liver steatosis in liver specimens from HCV-infected patients [429] and HCV-activated autophagy has been shown to degrade mitochondria [430]. Therefore, further study is needed to examine whether HCV-activated autophagy alters metabolic homeostasis and organelle regeneration, thus accelerating the progression of HCV-related metabolic diseases. On the other hand, autophagy has been demonstrated to be critical for the suppression of tumor development. The gene knockout of ATGs, such as Beclin 1 and ATG5, in mice has been shown to interfere with the autophagic process and promote the development of malignant tumors [447,448,449,450,451]. Furthermore, several studies have shown high p62/SQSTM1 expression in various types of HCC tissues and cell lines [450,452], suggesting that autophagy interference is a major route for the induction of hepatocarcinogenesis. HCV infection likely initiates an autophagic process to benefit viral growth, perhaps repressing basal autophagy to alter metabolic homeostasis and induce the development of liver cancer. Accordingly, a recent study implied that p62/SQSTM1 is highly phosphorylated at serine 349 in the tumor tissues of HCV-positive HCC patients and that phosphorylated p62/SQSTM1 drives the glucoronate and glutathione synthesis pathways to enhance drug resistance and malignancy of liver cancer [453]. Increasing evidence has demonstrated that tumor cells may trigger autophagy to circumvent the tumor microenvironment and thus increase cell proliferation potency [454,455,456,457,458,459,460,461] and counteract a variety of stress responses, such as hypoxia and nutrient deprivation [457,458,461]. Moreover, autophagy has also been shown to induce drug resistance in anticancer treatment [454,455,456]. Together, these studies suggest that HCV infection may activate autophagy to establish a cell microenvironment that favors cell surveillance under stress conditions in chronically infected patients, possibly promoting the development of HCV-associated end-stage liver diseases. Nevertheless, a feasible and small animal model that enables investigation of the complete HCV life cycle in the liver is urgently needed to uncover the impact of HCV-autophagy interactions on the progression of HCV-related liver diseases.
4.2. DENV
Analogous to HCV, DENV infection also activates host autophagy in infected cells [398,462,463,464,465]. Lee and colleagues first showed that DENV serotype 2 (DENV-2) infection induces autophagy in infected Huh7 cells, as demonstrated by the formation of autophagosomes and the increased level of LC3 lipidation (Table 2) [398]. The fact that genetically silencing ATG5 by RNA interference inhibited DENV replication implies the pro-viral effect of host autophagy on promoting DENV replication [398]. Soon afterward, Panyasrivanit et al. also reported that DENV-2 induces autophagy in viral-infected HepG2 cells (Table 2) [465]. The colocalization of dsRNA, DENV NS1 and ribosomal protein L28 within LC3-labeled autophagic vacuoles and the observation of mannose-6-phosphate receptor (MPR)-enriched amphisomes suggest that DENV-induced autophagy machinery functions as a platform for DENV replication [465,466]. Interference of autophagy initiation by 3-MA dramatically reduces the intracellular amount of viral RNA and extracellular virions [465,466], implying that the autophagic process is required for organization of the DENV replication compartment. In contrast, inhibition of autophagosome fusion with lysosomes by L-Asparagine (L-Asn) slightly increases the levels of intracellular viral RNA and extracellular virions [465,466], suggesting the potential role of autolysosomes in eliminating DENV. In addition to DENV-2, DENV-3 was also shown to induce autophagy to form autolysosomes that encompass cathepsin D, NS1 and dsRNA and support viral RNA replication in HepG2 cells (Table 2) [464], again arguing that DENV infection activates autophagy to reconstitute membranous structures for viral growth. In contrast, DENV-2 infection of U937 cells induces the accumulation of autophagosomes by interfering with their fusion with lysosomes (Table 2) [462]. Interruption of autophagy initiation by a dominant Vps34/PI3KC3 mutant decreases the replication of DENV viral RNA and the secretion of infectious DENV particles, indicating that DENV-2 infection also promotes autophagy to benefit the viral life cycle. Additionally, this study implies that DENV infection differentially modulates autophagy in different infectious cell models. The induction of autophagy by DENV infection was further confirmed by an in vivo study on DENV infection in mice (Table 2) [467]. In this study, Lee and colleagues demonstrated that DENV infection triggers the formation of autophagic vacuoles and the lipidation of LC3 in infected brain tissues (Table 2) [467]. Most importantly, the inhibition of autophagy in DENV-infected mice represses viral replication and the forwarded induction of DENV-induced autophagy by rapamycin promotes disease progression, supporting a new idea in which autophagy not only plays a pro-viral role in DENV growth but also participates in the pathogenesis of DENV-associated diseases.
Apart from viral RNA replication within autophagic machinery, the pharmacological modulation of autophagy in DENV-infected cells showed that DENV infection activates autophagy to facilitate the maturation of infectious particles (Table 2) [468]. Notably, autophagy was shown to facilitate DENV infection via the ADE-mediated pathway (Table 2) [469] and to associate with DENV infectious particles to escape antibody neutralization and promote cell-to-cell transmission [470]. Moreover, Chu et al. reported that autophagy interacts with DENV during the initial stage of infection to facilitate the DENV entry process (Table 2) [471]. Furthermore, Bartenschlager’s group demonstrated that DENV infection enhances autophagic flux to promote viral replication during the initial stage of infection and induces the degradation of p62/SQSTM1 via a proteasomal mechanism (Table 2) [472]. Together, these studies imply multiple roles of autophagy in the DENV life cycle.
In addition to playing a pro-viral role in the DENV viral life cycle, autophagy activated by DENV infection also alters cellular metabolism, as first demonstrated by Heaton et al. (Table 2) [463]. This study demonstrated that DENV triggers the autophagic process all the way through autolysosome maturation in various types of cells to catabolize LDs to release FFA and generate ATP for the replication of DENV [463]. Very recently, Zhang et al. reported that DENV NS4A interacts with ancient ubiquitous proteins 1 (AUP1), an LD-associated VLDL assembly regulator, to promote DENV-induced lipophagy, thus promoting virus production (Table 2) [473]. Another study demonstrated that DENV infection activates the AMPK-mTOR axis to promote lipophagy (Table 2) [474]. These results collectively conclude that DENV activates autophagy to promote LD catabolism, thus increasing the β-oxidation of FFA for viral replication.
Several studies have investigated how DENV infection induces autophagy activation and DENV NS4A has been shown to activate autophagy in a PI3K-dependent manner (Table 2) [475]. Additionally, NS1 and NS4B reportedly participate in DENV-induced autophagy, promoting the degradation of LDs and viral-induced vascular leakage (Table 2) [473,476]. In a similar fashion to HCV, DENV also triggers the ER stress/UPR pathway to activate autophagy, thus promoting viral replication and cell surveillance (Table 2) [477,478,479].
Although autophagy was extensively shown to be activated by DENV infection, little is known about how DENV initiates this process. Additionally, the exact physiological significance of viral-induced autophagy in DENV-host interactions and the pathogenesis of DENV-associated diseases remain under further investigation. Comprehensively understanding the functional roles of DENV-activated autophagy in host cells will not only help unveil the progression of DENV-related diseases but also provide a molecular basis for the rational design of antiviral treatment.
4.3. JEV
JEV infection was first reported to activate autophagy in human NT-2 cells (a pluripotent human testicular embryonal carcinoma cell line), as demonstrated by increased levels of lipidated LC3 (Table 3) [480]. This study also showed that induction of autophagy by rapamycin enhances JEV replication, whereas the autophagy inhibitor 3-MA suppresses viral growth [480]. Silencing of ATG5 and Beclin 1 genes in JEV-infected cells reduces viral replication, suggesting that JEV activates autophagy to benefit viral growth [480]. The lack of colocalization between dsRNA and LC3-labeled autophagic vacuoles suggests that JEV-induced autophagy machinery does not provide membrane compartments for JEV replication [480]. Moreover, the specific colocalization of LC3-labeled autophagic vacuoles with JEV during the initial stage of infection implies that viral-activated autophagy participates in the JEV entry process. Later, Jin et al. also demonstrated that JEV infection activates the entire autophagic process through autolysosome maturation, which is required for JEV replication (Table 3) [481]. Interference with autophagy by knocking down the ATG5 and Beclin 1 genes abolishes the replication of JEV viral RNA, accompanied by enhanced cell apoptosis and induced antiviral immunity [481]. On the other hand, the loss of p62/SQSTM1 in mouse embryonic fibroblasts downregulates the viral replication of JEV (Table 3) [482], suggesting that the p62/SQSTM1-mediated autophagic process promotes JEV growth in infected cells. Moreover, the C, PrM and NS3 proteins were shown to induce JEV-induced autophagy, presumably via IRGM (Table 3) [483]. Similar to HCV and DENV, the ER stress/UPR pathway is required for JEV-induced autophagy activation in neuronal cells (Table 3) [484]. A recent study reported that diphenyleneiodonium, an antioxidant drug, represses JEV-activated autophagy by inhibiting the ER/UPR pathway to suppress JEV production in neuronal Neuro2a cells (Table 3) [485], highlighting that interfering with JEV-activated UPR and autophagy might be a therapeutic target for inhibiting JEV infection. In contrast, JEV infection was also shown to attenuate autophagy to promote viral replication via the ubiquitin E3 ligase activity of Nedd4 and organization of the nonlipidated LC3-containing EDEM1 (ER degradation enhancer, mannosidase α-like 1)-associated membrane (Table 3) [486,487]. Thus far, little is known about whether JEV-activated autophagy participates in the progression of neurological diseases but this information could be obtained by investigating how JEV induces autophagy and comprehensively understanding the detailed functions of autophagy in the JEV viral life cycle.
4.4. WNV
In contrast to the pro-viral role of autophagy in DENV and JEV, autophagy is not likely required for activation of the WNV viral life cycle. WNV infection was shown to induce LC3 lipidation and the formation of LC3-labeled autophagic vacuoles in Vero cells (Table 4) [488]. The loss of ATG5 and related autophagy by knockdown and knockout methods, respectively, did not alter WNV replication in infected cells [488], suggesting that WNV-activated autophagy is not required for the infectious life cycle of WNV. This conclusion is further supported by other studies showing that depletion of ATG5 and ATG7 has no significant effect on the production of WNV infectious particles (Table 4) [489,490] and the supporting role of mTOR signaling for the translation of WNV viral RNA (Table 4) [491]. These studies provided a mechanistic basis for developing an autophagy inducer, Tat-Beclin 1, to inhibit WNV replication and disease progression in WNV-infected mice (Table 4) [490,492]. Unlike DENV and JEV, the UPR is not coupled or required for the activation of WNV-induced autophagy [493]. Collectively, these studies suggest that autophagy may play an antiviral role in restricting WNV infection. However, whether autophagy participates in the pathogenesis of WNV-associated neurological diseases remains unknown and successfully answering this question will facilitate the design of a feasible and effective antiviral therapy against WNV infection.
4.5. ZIKV
In recent years, Zika was discovered to activate autophagy to induce the formation of autophagosomes and thus promote viral replication in human skin cells (Table 5) [494]. Additionally, the modulation of autophagy by pharmacological regulators is associated with ZIKV replication in infected cells [494], supporting the notion that autophagy plays a pro-viral role in ZIKV infection. Moreover, ZIKV NS4A and NS4B were demonstrated to induce autophagy in human fetal neural stem cells (fNSCs) by deregulating Akt-mTOR signaling (Table 5) [495]. This Zika NS4A- and NS4B-mediated autophagy activation is required for ZIKV replication and the impaired neurogenesis of fNSCs [495]. Very recently, ZIKV infection of human umbilical vein endothelial cells (HUVECs) was shown to induce complete autophagy and thus promote p62/SQSTM1 degradation, which is critically required for ZIKV replication (Table 5) [496]. These studies indicated that ZIKV-activated autophagy may participate in the pathogenesis of ZIKV-related diseases and suggest that the inhibition of ZIKV-induced autophagy may represent a therapeutic target for inhibiting ZIKV infection. This hypothesis is further supported by a recent study showing that deletion of ATG16L and treatment with chloroquine, an inhibitor of autolysosome maturation, restricts ZIKV vertical transmission in pregnant mice and ameliorates the placental and fetal outcomes of ZIKV infection (Table 5) [497]. However, recent studies indicated that inflammation-induced autophagy limits ZIKV infection via the stimulator of interferon genes (STING) in the Drosophila brain (Table 5) [498], implying that ZIKV-activated autophagy may represent a new route for eliminating ZIKV infection via inflammation. The physiological significance of autophagy in the life cycle and transmission of ZIKV as well as the mechanisms underlying the pathogenesis of ZIKV-induced neonatal disorders continue to be elucidated and this information is urgently required for development of an effective anti-ZIKV drug for clinical use.
5. Autophagy as a Therapeutic Target for Curing Flaviviruses Infection
Suppression of autophagy has been demonstrated to inhibit viral growth in most of flavivirus-infected cells. Interference with membrane nucleation by the Vps34/PI3KC3 inhibitor, 3-MA reduces the viral replications of DENV [398,462,463,464,465,466,467,471], JEV [480,483], WNV [488] and ZIKV [497]. It is noted that 3-MA increases the survival rate and mitigates the symptoms of DENV-infected mice [467,468], implying that autophagy inhibition can be used to control disease progress in DENV infection in vivo. Inhibition of maturation and activity of autolysosomes by chloroquine and bafilomycin-A1 also has an inhibitory effect on virus growth in HCV [497,499], DENV [500,501,502,503], JEV [504,505] and ZIKV [495,496,497,506,507,508]. In addition, chloroquine was also shown to mitigate DENV-related symptoms in patients [509,510,511] and inhibit the maternal-fetal transmission of ZIKV and protect fetal mice from microcephaly caused by ZIKV infection [497,507,508]. Moreover, spautin-1 was also shown to reduce the maturation of DENV virions [468]. These studies collectively indicate that the inhibition of host autophagy may present a feasible strategy of restricting flavivirus infection. However, deregulated autophagy in some cases of flaviviral infection leads to the cell death of infected cells, such as HCV [407,418] and JEV [481,487], suggesting a risk of unexpected tissue damage by viral infection, which could be caused by a defect in the cell survival function of autophagy. In contrast, induction of autophagy by Tat-Beclin 1 suppress WNV infection in vitro and in vivo [490,492], implying that induction of autophagy may promote the xenophagic elimination of infectious WNV. Although modulation of autophagy, particularly the inhibition of autophagy can be conceivably utilized to combat flaviviral infection, the specific autophagy inhibitors that contain higher potency and lower cytotoxicity still remain to be discovered. Given that the roles of autophagy in flavivirus infection is context- and disease-dependent, the impacts of autophagy inhibitors in the cytoprotective and pro-survival functions of autophagy should be investigated before the use of these strategies in clinical medicine [5,6,512,513]. Interference with host autophagy may provide for virus infection control but may also deregulate metabolism homeostasis and disrupt the elimination of damaged materials caused by viral infection in host cells. For instance, the inhibition of HCV infection by the repression of autophagy may introduce the risk of developing liver-related diseases since autophagy defects have been shown to induce several types of diseases in liver [514,515]. Before we attempt to inhibit autophagy for therapeutic intervention in flavivirus infection, a deeper and more comprehensive understanding of flaviviral viruses and host interactions is needed. In addition, the different types of treatment strategies that are disease progress dependent and the improved safety of drugs for clinic use are urgently required.
6. Conclusions and Future Directions
Autophagy plays a pivotal role in balancing cellular homeostasis via the continuous degradation of intracellular components and recycling of nutrients for biogenesis. Upon the infection of cells with flavivirus, autophagy is exploited by the virus to reconstitute the membrane structure required for completion of the viral life cycle and degrade harmful organelles in the infected cells. Additionally, autophagy is activated to allow flaviviruses to escape innate immune surveillance and protect infected cells from death. Although the interactions between flaviviruses and host autophagy have been extensively studied over the past decade, several fundamental questions regarding how flaviviruses activate autophagy and how viral-induced autophagy influences the pathogeneses of flavivirus-related diseases remain unknown. Future investigations are needed to understand the clinical relevance of autophagy and flavivirus infections in infected patients. For instance, whether the conclusions drawn from these in vitro infected cell models can be recapitalized in real in vivo physiological conditions needs to be determined. Additionally, the lack of a feasible and conceivable mouse model for most flaviviruses substantially hampers the development of a dynamic and timely assay for assessing flavivirus-induced autophagy in vivo. Dissecting the interplay between flaviviruses and autophagy in an in vivo animal that supports the complete life cycle will permit the detailed elucidation of the functional roles of autophagy in viral infection, host defense regulation and disease progression. An innovative high-throughput screening strategy and bioinformatics analysis tools will improve the comprehensive understanding of autophagy-flavivirus interactions and help identify potential autophagy therapeutic targets to design an effective antiviral strategy for curing flavivirus infection in the clinic.
Author Contributions
Conceptualization, P.-Y.K.; Writing-Original Draft Preparation, P.-Y.K.; Writing-Review & Editing, P.-Y.K.
Funding
This study was supported by research grants from the Ministry of Science and Technology (MOST 102-2320-B-182-037-MY3 and MOST 105-2628-B-182-001-MY3), Taipei, National Health Research Institute (NHRI-EX103-10322SC, NHRI-EX104-10322SC, NHRI-EX105-10322SC and NHRI-EX106-10322SC), Miaoli and Chang Gung Memorial Hospital (CMRPD1C0211, CMRPD1D0021, CMRPD1D0022, CMRPD1D0023, CMRPD1G0281, CRRPD1F0031 and CMRPG3F0571), Taoyuan, Taiwan.
Acknowledgments
The author apologizes to the colleagues whose work could not be referenced due to space constraints.
Conflicts of Interest
The author declares no conflict of interest.
References
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Rozieres, A.; Faure, M. Autophagy during Early Virus-Host Cell Interactions. J. Mol. Biol. 2018, 430, 1696–1713. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Vicenzi, E.; Pagani, I.; Ghezzi, S.; Taylor, S.L.; Rudd, T.R.; Lima, M.A.; Skidmore, M.A.; Yates, E.A. Subverting the mechanisms of cell death: Flavivirus manipulation of host cell responses to infection. Biochem. Soc. Trans. 2018, 46, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.L., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B.; Essner, E. Cytolysomes and mitochondrial degeneration. J. Cell Biol. 1962, 15, 140–146. [Google Scholar] [CrossRef]
- Novikoff, A.B. The proximal tubule cell in experimental hydronephrosis. J. Biophys. Biochem. Cytol. 1959, 6, 136–138. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Essner, E. Pathological changes in cytoplasmic organelles. Fed. Proc. 1962, 21, 1130–1142. [Google Scholar]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef]
- Pfeifer, U. Inhibition by insulin of the physiological autophagic breakdown of cell organelles. Acta Biol. Med. Ger. 1977, 36, 1691–1694. [Google Scholar] [PubMed]
- Pfeifer, U.; Strauss, P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J. Mol. Cell. Cardiol. 1981, 13, 37–49. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Ward, W.F. Behavior of the lysosomal system during organ perfusion. An inquiry into the mechanism of hepatic proteolysis. Front. Biol. 1976, 45, 157–184. [Google Scholar] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holen, I.; Gordon, P.B.; Seglen, P.O. Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem. J. 1992, 284, 633–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houri, J.J.; Ogier-Denis, E.; De Stefanis, D.; Bauvy, C.; Baccino, F.M.; Isidoro, C.; Codogno, P. Differentiation-dependent autophagy controls the fate of newly synthesized N-linked glycoproteins in the colon adenocarcinoma HT-29 cell line. Biochem. J. 1995, 309, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankson, H.; Holen, I.; Seglen, P.O. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp. Cell Res. 1995, 218, 522–530. [Google Scholar] [CrossRef]
- Holen, I.; Stromhaug, P.E.; Gordon, P.B.; Fengsrud, M.; Berg, T.O.; Seglen, P.O. Inhibition of autophagy and multiple steps in asialoglycoprotein endocytosis by inhibitors of tyrosine protein kinases (tyrphostins). J. Biol. Chem. 1995, 270, 12823–12831. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Luiken, J.J.; Blommaart, P.J.; van Woerkom, G.M.; Meijer, A.J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995, 270, 2320–2326. [Google Scholar] [CrossRef]
- Meijer, A.J. Inhibition of autophagic proteolysis by cell swelling in hepatocytes. Biochem. J. 1995, 312, 987–988. [Google Scholar] [CrossRef] [Green Version]
- Fengsrud, M.; Roos, N.; Berg, T.; Liou, W.; Slot, J.W.; Seglen, P.O. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: Effects of vinblastine and asparagine on vacuole distributions. Exp. Cell Res. 1995, 221, 504–519. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Couvineau, A.; Maoret, J.J.; Houri, J.J.; Bauvy, C.; De Stefanis, D.; Isidoro, C.; Laburthe, M.; Codogno, P. A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J. Biol. Chem. 1995, 270, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.B.; Kovacs, A.L.; Seglen, P.O. Temperature dependence of protein degradation, autophagic sequestration and mitochondrial sugar uptake in rat hepatocytes. Biochim. Biophys. Acta 1987, 929, 128–133. [Google Scholar] [CrossRef]
- Gordon, P.B.; Seglen, P.O. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 1988, 151, 40–47. [Google Scholar] [CrossRef]
- Plomp, P.J.; Wolvetang, E.J.; Groen, A.K.; Meijer, A.J.; Gordon, P.B.; Seglen, P.O. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 1987, 164, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Matsuura, A.; Wada, Y.; Ohsumi, Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1995, 210, 126–132. [Google Scholar] [CrossRef]
- Harding, T.M.; Hefner-Gravink, A.; Thumm, M.; Klionsky, D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996, 271, 17621–17624. [Google Scholar] [CrossRef]
- Schlumpberger, M.; Schaeffeler, E.; Straub, M.; Bredschneider, M.; Wolf, D.H.; Thumm, M. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 1068–1076. [Google Scholar] [CrossRef]
- Straub, M.; Bredschneider, M.; Thumm, M. AUT3, a serine/threonine kinase gene, is essential for autophagocytosis in Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 3875–3883. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Schaeffeler, E.; Bernreuther, D.; Bredschneider, M.; Wolf, D.H.; Thumm, M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998, 17, 3597–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Formation and maturation of autophagosomes in higher eukaryotes: A social network. Curr. Opin. Cell Biol. 2018, 53, 29–36. [Google Scholar] [CrossRef]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Johansen, T. FYCO1: Linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010, 6, 550–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef] [Green Version]
- Fracchiolla, D.; Sawa-Makarska, J.; Martens, S. Beyond Atg8 binding: The role of AIM/LIR motifs in autophagy. Autophagy 2017, 13, 978–979. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Sawa-Makarska, J.; Zens, B.; Ruiter, A.; Zaffagnini, G.; Brezovich, A.; Romanov, J.; Runggatscher, K.; Kraft, C.; Zagrovic, B.; et al. Mechanism of cargo-directed Atg8 conjugation during selective autophagy. Elife 2016, 5, e18544. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Oikawa, K.; Yoshimoto, K.; Kondo, M.; Mano, S.; Yamada, K.; Hayashi, M.; Sakamoto, W.; Ohsumi, Y.; Nishimura, M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013, 25, 4967–4983. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Tan, L.J.; Mochida, K.; Kirisako, H.; Koizumi, M.; Asai, E.; Sakoh-Nakatogawa, M.; Ohsumi, Y.; Nakatogawa, H. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J. Cell Biol. 2014, 207, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [Green Version]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Liu, Y.; Schiff, M.; Czymmek, K.; Talloczy, Z.; Levine, B.; Dinesh-Kumar, S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005, 121, 567–577. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Gregoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Tanaka, K.; Komatsu, M. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Lett. 2014, 588, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Munz, C. Enhancing immunity through autophagy. Annu. Rev. Immunol. 2009, 27, 423–449. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Eating the enemy within: Autophagy in infectious diseases. Cell Death Differ. 2009, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sowers, J.R.; Zhang, Y. Metabolic Stress, Autophagy, and Cardiovascular Aging: From Pathophysiology to Therapeutics. Trends Endocrinol. Metab. 2018, 29, 699–711. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef]
- Valenzuela, J.G.; Aksoy, S. Impact of vector biology research on old and emerging neglected tropical diseases. PLoS Negl. Trop. Dis. 2018, 12, e0006365. [Google Scholar] [CrossRef]
- Shaw, W.R.; Catteruccia, F. Vector biology meets disease control: Using basic research to fight vector-borne diseases. Nat. Microbiol. 2018. [Google Scholar] [CrossRef]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Diagana, M.; Preux, P.M.; Dumas, M. Japanese encephalitis revisited. J. Neurol. Sci. 2007, 262, 165–170. [Google Scholar] [CrossRef]
- Maximova, O.A.; Pletnev, A.G. Flaviviruses and the Central Nervous System: Revisiting Neuropathological Concepts. Annu. Rev. Virol. 2018, 5, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Hernandez-Triana, L.M.; Banyard, A.C.; Fooks, A.R.; Johnson, N. Japanese encephalitis virus infection, diagnosis and control in domestic animals. Vet. Microbiol. 2017, 201, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Ngono, A.E.; Shresta, S. Immune Response to Dengue and Zika. Annu. Rev. Immunol. 2018, 36, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Turtle, L.; Solomon, T. Japanese encephalitis—The prospects for new treatments. Nat. Rev. Neurol. 2018, 14, 298–313. [Google Scholar] [CrossRef]
- Patel, H.; Sander, B.; Nelder, M.P. Long-term sequelae of West Nile virus-related illness: A systematic review. Lancet Infect. Dis. 2015, 15, 951–959. [Google Scholar] [CrossRef]
- Luo, H.; Wang, T. Recent advances in understanding West Nile virus host immunity and viral pathogenesis. F1000Res 2018, 7, 338. [Google Scholar] [CrossRef]
- Gack, M.U.; Diamond, M.S. Innate immune escape by Dengue and West Nile viruses. Curr. Opin. Virol. 2016, 20, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Russo, F.B.; Jungmann, P.; Beltrao-Braga, P.C.B. Zika infection and the development of neurological defects. Cell. Microbiol. 2017, 19, 949–958. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Faizan, M.I.; Abdullah, M.; Ali, S.; Naqvi, I.H.; Ahmed, A.; Parveen, S. Zika Virus-Induced Microcephaly and Its Possible Molecular Mechanism. Intervirology 2016, 59, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.X.; Stiasny, K. Flaviviruses and flavivirus vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Yamanaka, A.; Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 2014, 32, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-acting Antiviral Therapy for HCV Infection and Remaining Challenges. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Boldescu, V.; Behnam, M.A.M.; Vasilakis, N.; Klein, C.D. Broad-spectrum agents for flaviviral infections: Dengue, Zika and beyond. Nat. Rev. Drug Discov. 2017, 16, 565–586. [Google Scholar] [CrossRef]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V.; Zuckerman, A.J. Transfusion-associated hepatitis not due to viral hepatitis type A or B. Rev. Med. Virol. 2001, 11, 3–8. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Chayama, K.; Hayes, C.N. Hepatitis C virus: How genetic variability affects pathobiology of disease. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roingeard, P. Hepatitis C virus diversity and hepatic steatosis. J. Viral Hepat. 2013, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Hong, F.; Radaeva, S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology 2004, 39, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.A.; Chung, R.T. The impact of human gene polymorphisms on HCV infection and disease outcome. Semin. Liver Dis. 2011, 31, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Gigi, E.; Lagopoulos, V.I.; Bekiari, E. Hepatocellular carcinoma occurrence in DAA-treated hepatitis C virus patients: Correlated or incidental? A brief review. World J. Hepatol. 2018, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Sessa, A.; Cossiga, V.; Morando, F.; Caporaso, N.; Morisco, F. Direct-acting antivirals and hepatocellular carcinoma in chronic hepatitis C: A few lights and many shadows. World J. Gastroenterol. 2018, 24, 2582–2595. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology 2016, 64, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Green, P.K.; Berry, K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J. Hepatol. 2017. [Google Scholar] [CrossRef]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A. Liver cancer: Effect of HCV clearance with direct-acting antiviral agents on HCC. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar]
- Suzuki, T. Assembly of hepatitis C virus particles. Microbiol. Immunol. 2011, 55, 12–18. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- McKeating, J.A.; Zhang, L.Q.; Logvinoff, C.; Flint, M.; Zhang, J.; Yu, J.; Butera, D.; Ho, D.D.; Dustin, L.B.; Rice, C.M.; et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 2004, 78, 8496–8505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef]
- Xu, Y.; Martinez, P.; Seron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, S.; Zhang, J.; Baribaud, F.; Chen, Z.; Leslie, G.J.; Lin, G.; Granelli-Piperno, A.; Doms, R.W.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.K.; Grove, J.; Hu, K.; Mee, C.J.; Balfe, P.; McKeating, J.A. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J. Virol. 2009, 83, 12407–12414. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Dubuisson, J. Hepatitis C virus entry into host cells. Cell. Mol. Life Sci. 2008, 65, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Park, S.; Yang, J.S.; Kwon, O.S.; Kim, S.; Jang, S.K. Discovery of cellular proteins required for the early steps of HCV infection using integrative genomics. PLoS ONE 2013, 8, e60333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lee, E.M.; Hammack, C.; Robotham, J.M.; Basu, M.; Lang, J.; Brinton, M.A.; Tang, H. Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J. Virol. 2014, 88, 8433–8444. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Yuan, F.; Li, Y.; Zhu, F.; Hou, P.; Li, J.; Song, X.; Ding, M.; Deng, H. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 2007, 81, 12465–12471. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H. Hepatitis C: The clinical spectrum of disease. Hepatology 1997, 26, S15–S20. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, S21–S29. [Google Scholar] [Green Version]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bisceglie, A.M.; Goodman, Z.D.; Ishak, K.G.; Hoofnagle, J.H.; Melpolder, J.J.; Alter, H.J. Long-term clinical and histopathological follow-up of chronic posttransfusion hepatitis. Hepatology 1991, 14, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M.; Order, S.E.; Klein, J.L.; Waggoner, J.G.; Sjogren, M.H.; Kuo, G.; Houghton, M.; Choo, Q.L.; Hoofnagle, J.H. The role of chronic viral hepatitis in hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 1991, 86, 335–338. [Google Scholar] [PubMed]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Villano, S.A.; Vlahov, D.; Nelson, K.E.; Cohn, S.; Thomas, D.L. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology 1999, 29, 908–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnello, V.; Chung, R.T.; Kaplan, L.M. A role for hepatitis C virus infection in type II cryoglobulinemia. N. Engl. J. Med. 1992, 327, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Charles, E.D.; Dustin, L.B. Hepatitis C virus-induced cryoglobulinemia. Kidney Int. 2009, 76, 818–824. [Google Scholar] [CrossRef]
- Clement, S.; Pascarella, S.; Negro, F. Hepatitis C virus infection: Molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses 2009, 1, 126–143. [Google Scholar] [CrossRef]
- Negro, F.; Alaei, M. Hepatitis C virus and type 2 diabetes. World J. Gastroenterol. 2009, 15, 1537–1547. [Google Scholar] [CrossRef]
- Guzman, M.G.; Kouri, G. Dengue: An update. Lancet Infect. Dis. 2002, 2, 33–42. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Coloma, J.; Harris, E. Dengue: Knowledge gaps, unmet needs, and research priorities. Lancet Infect. Dis. 2017, 17, e88–e100. [Google Scholar] [CrossRef]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hotta, S. Experimental studies on dengue. I. Isolation, identification and modification of the virus. J. Infect. Dis. 1952, 90, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Ni, H.; Xu, R.; Barrett, A.D.; Watowich, S.J.; Gubler, D.J.; Weaver, S.C. Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J. Virol. 2000, 74, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.L.; Harris, E. Global spread and persistence of dengue. Annu. Rev. Microbiol. 2008, 62, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengue virus-mosquito interactions. Ann. Rev. Entomol. 2008, 53, 273–291. [Google Scholar] [CrossRef]
- Guzman, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martinez, E.; et al. Dengue: A continuing global threat. Nat. Rev. Microbiol. 2010, 8, S7–S16. [Google Scholar] [CrossRef]
- Simmons, C.P.; McPherson, K.; Van Vinh Chau, N.; Hoai Tam, D.T.; Young, P.; Mackenzie, J.; Wills, B. Recent advances in dengue pathogenesis and clinical management. Vaccine 2015, 33, 7061–7068. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef]
- Cruz-Oliveira, C.; Freire, J.M.; Conceicao, T.M.; Higa, L.M.; Castanho, M.A.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef] [Green Version]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef]
- Van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Zaitseva, E.; Yang, S.T.; Melikov, K.; Pourmal, S.; Chernomordik, L.V. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010, 6, e1001131. [Google Scholar] [CrossRef]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Despres, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef]
- Miller, J.L.; de Wet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Chen, Y.C.; Wang, S.Y.; King, C.C. Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/macrophages by blockade of virus entry via a CD14-dependent mechanism. J. Virol. 1999, 73, 2650–2657. [Google Scholar]
- Reyes-Del Valle, J.; Chavez-Salinas, S.; Medina, F.; Del Angel, R.M. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 2005, 79, 4557–4567. [Google Scholar] [CrossRef]
- Jindadamrongwech, S.; Thepparit, C.; Smith, D.R. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 2004, 149, 915–927. [Google Scholar] [CrossRef]
- Aoki, C.; Hidari, K.I.; Itonori, S.; Yamada, A.; Takahashi, N.; Kasama, T.; Hasebe, F.; Islam, M.A.; Hatano, K.; Matsuoka, K.; et al. Identification and characterization of carbohydrate molecules in mammalian cells recognized by dengue virus type 2. J. Biochem. 2006, 139, 607–614. [Google Scholar] [CrossRef]
- Wichit, S.; Jittmittraphap, A.; Hidari, K.I.; Thaisomboonsuk, B.; Petmitr, S.; Ubol, S.; Aoki, C.; Itonori, S.; Morita, K.; Suzuki, T.; et al. Dengue virus type 2 recognizes the carbohydrate moiety of neutral glycosphingolipids in mammalian and mosquito cells. Microbiol. Immunol. 2011, 55, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Thepparit, C.; Smith, D.R. Serotype-specific entry of dengue virus into liver cells: Identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J. Virol. 2004, 78, 12647–12656. [Google Scholar] [CrossRef]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef]
- Gao, F.; Duan, X.; Lu, X.; Liu, Y.; Zheng, L.; Ding, Z.; Li, J. Novel binding between pre-membrane protein and claudin-1 is required for efficient dengue virus entry. Biochem. Biophys. Res. Commun. 2010, 391, 952–957. [Google Scholar] [CrossRef]
- Che, P.; Tang, H.; Li, Q. The interaction between claudin-1 and dengue viral prM/M protein for its entry. Virology 2013, 446, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. MBio 2016, 7, e00804-16. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Miller, S. Molecular aspects of Dengue virus replication. Future Microbiol. 2008, 3, 155–165. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Flavivirus modulation of cellular metabolism. Curr. Opin. Virol. 2016, 19, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: The enemy inside—Caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Schuh, A.J.; Ward, M.J.; Brown, A.J.; Barrett, A.D. Phylogeography of Japanese encephalitis virus: Genotype is associated with climate. PLoS Negl. Trop. Dis. 2013, 7, e2411. [Google Scholar] [CrossRef]
- Lewis, L.; Taylor, H.G.; Sorem, M.B.; Norcross, J.W.; Kindsvatter, V.H. Japanese B encephalitis; clinical observations in an outbreak on Okinawa Shima. Arch. Neurol. Psychiatry 1947, 57, 430–463. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, J.M.; Song, B.H.; Yun, S.I.; Yun, G.N.; Byun, S.J.; Lee, Y.M. Profiling of viral proteins expressed from the genomic RNA of Japanese encephalitis virus using a panel of 15 region-specific polyclonal rabbit antisera: Implications for viral gene expression. PLoS ONE 2015, 10, e0124318. [Google Scholar] [CrossRef]
- Stocks, C.E.; Lobigs, M. Signal peptidase cleavage at the flavivirus C-prM junction: Dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 1998, 72, 2141–2149. [Google Scholar]
- Yun, S.I.; Lee, Y.M. Early Events in Japanese Encephalitis Virus Infection: Viral Entry. Pathogens 2018, 7, 68. [Google Scholar] [CrossRef]
- Nain, M.; Abdin, M.Z.; Kalia, M.; Vrati, S. Japanese encephalitis virus invasion of cell: Allies and alleys. Rev. Med. Virol. 2016, 26, 129–141. [Google Scholar] [CrossRef]
- Chiou, S.S.; Liu, H.; Chuang, C.K.; Lin, C.C.; Chen, W.J. Fitness of Japanese encephalitis virus to Neuro-2a cells is determined by interactions of the viral envelope protein with highly sulfated glycosaminoglycans on the cell surface. J. Med. Virol. 2005, 76, 583–592. [Google Scholar] [CrossRef]
- Su, C.M.; Liao, C.L.; Lee, Y.L.; Lin, Y.L. Highly sulfated forms of heparin sulfate are involved in japanese encephalitis virus infection. Virology 2001, 286, 206–215. [Google Scholar] [CrossRef]
- Liu, H.; Chiou, S.S.; Chen, W.J. Differential binding efficiency between the envelope protein of Japanese encephalitis virus variants and heparan sulfate on the cell surface. J. Med. Virol. 2004, 72, 618–624. [Google Scholar] [CrossRef]
- Mason, C.P.; Tarr, A.W. Human lectins and their roles in viral infections. Molecules 2015, 20, 2229–2271. [Google Scholar] [CrossRef]
- Kim, J.M.; Yun, S.I.; Song, B.H.; Hahn, Y.S.; Lee, C.H.; Oh, H.W.; Lee, Y.M. A single N-linked glycosylation site in the Japanese encephalitis virus prM protein is critical for cell type-specific prM protein biogenesis, virus particle release, and pathogenicity in mice. J. Virol. 2008, 82, 7846–7862. [Google Scholar] [CrossRef]
- Chen, S.T.; Liu, R.S.; Wu, M.F.; Lin, Y.L.; Chen, S.Y.; Tan, D.T.; Chou, T.Y.; Tsai, I.S.; Li, L.; Hsieh, S.L. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog. 2012, 8, e1002655. [Google Scholar] [CrossRef]
- Liu, K.; Qian, Y.; Jung, Y.S.; Zhou, B.; Cao, R.; Shen, T.; Shao, D.; Wei, J.; Ma, Z.; Chen, P.; et al. mosGCTL-7, a C-Type Lectin Protein, Mediates Japanese Encephalitis Virus Infection in Mosquitoes. J. Virol. 2017, 91, JVI-01348. [Google Scholar] [CrossRef]
- Fan, W.; Qian, P.; Wang, D.; Zhi, X.; Wei, Y.; Chen, H.; Li, X. Integrin alphavbeta3 promotes infection by Japanese encephalitis virus. Res. Vet. Sci. 2017, 111, 67–74. [Google Scholar] [CrossRef]
- Chuang, C.K.; Yang, T.H.; Chen, T.H.; Yang, C.F.; Chen, W.J. Heat shock cognate protein 70 isoform D is required for clathrin-dependent endocytosis of Japanese encephalitis virus in C6/36 cells. J. Gen. Virol. 2015, 96, 793–803. [Google Scholar] [CrossRef]
- Das, S.; Laxminarayana, S.V.; Chandra, N.; Ravi, V.; Desai, A. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology 2009, 385, 47–57. [Google Scholar] [CrossRef]
- Zhu, Y.Z.; Cao, M.M.; Wang, W.B.; Wang, W.; Ren, H.; Zhao, P.; Qi, Z.T. Association of heat-shock protein 70 with lipid rafts is required for Japanese encephalitis virus infection in Huh7 cells. J. Gen. Virol. 2012, 93, 61–71. [Google Scholar] [CrossRef]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91, JVI-02274. [Google Scholar] [CrossRef]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef]
- Liang, J.J.; Yu, C.Y.; Liao, C.L.; Lin, Y.L. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell. Microbiol. 2011, 13, 1358–1370. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Ravi, V.; Desai, A. Japanese encephalitis virus interacts with vimentin to facilitate its entry into porcine kidney cell line. Virus Res. 2011, 160, 404–408. [Google Scholar] [CrossRef]
- Hegde, N.R.; Gore, M.M. Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccin Immunother. 2017, 13, 1–18. [Google Scholar] [CrossRef]
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile virus: Review of the literature. JAMA 2013, 310, 308–315. [Google Scholar] [CrossRef]
- Francis, T.; Moore, A.E. A Study of the Neurotropic Tendency in Strains of the Virus of Epidemic Influenza. J. Exp. Med. 1940, 72, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. New insights into innate immune restriction of West Nile virus infection. Curr. Opin. Virol. 2015, 11, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Hall, R.A.; Lobigs, M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Nguyen, H.Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.E.; Koraka, P.; van den Doel, P.; Rimmelzwaan, G.F.; Haagmans, B.L.; Osterhaus, A.D. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008, 135, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Mattei, L.M.; Nguyen, H.Y.; Ansarah-Sobrinho, C.; Doms, R.W.; Pierson, T.C. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J. Biol. Chem. 2006, 291, 37183–37194. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.; Ng, M.L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 2004, 279, 54533–54541. [Google Scholar] [CrossRef]
- Lee, J.W.; Chu, J.J.; Ng, M.L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar] [CrossRef]
- Garcia, M.N.; Hasbun, R.; Murray, K.O. Persistence of West Nile virus. Microbes Infect. 2015, 17, 163–168. [Google Scholar] [CrossRef]
- Macnamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Morrison, T.E.; Diamond, M.S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J. Virol. 2017, 91, e00009-17. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Zhao, J.; Liu, X.; Fraser, K.; Lin, L.; Zhang, X.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 2017, 56, 1151–1162. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Aubry, M.; Teissier, A.; Huart, M.; Merceron, S.; Vanhomwegen, J.; Roche, C.; Vial, A.L.; Teururai, S.; Sicard, S.; Paulous, S.; et al. Zika Virus Seroprevalence, French Polynesia, 2014–2015. Emerg. Infect. Dis. 2017, 23, 669–672. [Google Scholar] [CrossRef]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome—Case report, French Polynesia, December 2013. Euro Surveill 2014, 19, 20720. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jimenez-Arango, J.A.; Zea-Vera, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Langerak, T.; Yang, H.; Baptista, M.; Doornekamp, L.; Kerkman, T.; Codrington, J.; Roosblad, J.; Vreden, S.G.; De Bruin, E.; Mogling, R.; et al. Zika Virus Infection and Guillain-Barre Syndrome in Three Patients from Suriname. Front. Neurol. 2016, 7, 233. [Google Scholar] [CrossRef]
- Ellul, M.A.; Soares, C.N.; Solomon, T. Zika virus and Guillain-Barre syndrome. J. R. Coll. Phys. Edinb. 2016, 46, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Do Rosario, M.S.; de Jesus, P.A.; Vasilakis, N.; Farias, D.S.; Novaes, M.A.; Rodrigues, S.G.; Martins, L.C.; Vasconcelos, P.F.; Ko, A.I.; Alcantara, L.C.; et al. Guillain-Barre Syndrome After Zika Virus Infection in Brazil. Am. J. Trop. Med. Hyg. 2016, 95, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Dirlikov, E.; Major, C.G.; Mayshack, M.; Medina, N.; Matos, D.; Ryff, K.R.; Torres-Aponte, J.; Alkis, R.; Munoz-Jordan, J.; Colon-Sanchez, C.; et al. Guillain-Barre Syndrome During Ongoing Zika Virus Transmission—Puerto Rico, January 1–July 31, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Wiwanitkit, V. Guillain-Barre syndrome and Zika virus infection. Arq. Neuropsiquiatr. 2016, 74, 692. [Google Scholar] [CrossRef] [PubMed]
- Bautista, L.E.; Sethi, A.K. Association between Guillain-Barre syndrome and Zika virus infection. Lancet 2016, 387, 2599–2600. [Google Scholar] [CrossRef]
- Das Neves Almeida, R.; Racine, T.; Magalhaes, K.G.; Kobinger, G.P. Zika Virus Vaccines: Challenges and Perspectives. Vaccines 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Stephenson, K.E.; Barouch, D.H. Zika virus vaccines. Nat. Rev. Microbiol. 2018, 16, 594–600. [Google Scholar] [CrossRef]
- Masmejan, S.; Baud, D.; Musso, D.; Panchaud, A. Zika virus, vaccines, and antiviral strategies. Expert Rev. Anti Infect. Ther. 2018, 16, 471–483. [Google Scholar] [CrossRef]
- Diamond, M.S.; Coyne, C.B. Vaccines in 2017: Closing in on a Zika virus vaccine. Nat. Rev. Immunol. 2018, 18, 89–90. [Google Scholar] [CrossRef]
- Richner, J.M.; Diamond, M.S. Zika virus vaccines: Immune response, current status, and future challenges. Curr. Opin. Immunol. 2018, 53, 130–136. [Google Scholar] [CrossRef]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron Microscopic Study of the Formation of Poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Burch, G.E.; Harb, J.M. Electron microscopic studies of viral pancreatitis in coxsackie B4 virus infected mice. Exp. Mol. Pathol. 1979, 31, 23–35. [Google Scholar] [CrossRef]
- Harb, J.M.; Burch, G.E. Spherical aggregates of coxsackie B4 virus particles in mouse pancreas. Beitr. Pathol. 1975, 156, 122–127. [Google Scholar] [CrossRef]
- Smith, J.D.; de Harven, E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J. Virol. 1978, 26, 102–109. [Google Scholar] [PubMed]
- Fine, D.L.; Lake, R.S.; Ludwig, E.H. Host-cell lysosomal response to two strains of herpes simplex virus. J. Virol. 1970, 5, 226–229. [Google Scholar] [PubMed]
- Nii, S.; Yasuda, I. Electron microscopic observations of FL cells infected with herpes simplex virus. III. Dense bodies. Biken J. 1976, 19, 53–61. [Google Scholar] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005, 3, e156. [Google Scholar] [CrossRef]
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 2006, 349, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey-Jurado, E.; Riedel, C.A.; Gonzalez, P.A.; Bueno, S.M.; Kalergis, A.M. Contribution of autophagy to antiviral immunity. FEBS Lett. 2015, 589, 3461–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumpter, R., Jr.; Levine, B. Selective autophagy and viruses. Autophagy 2011, 7, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.T.; Lee, J.; Narula, M.; Ou, J.J. Suppression of Host Innate Immune Response by Hepatitis C Virus via Induction of Autophagic Degradation of TRAF6. J. Virol. 2016, 90, 10928–10935. [Google Scholar] [CrossRef] [Green Version]
- Estrabaud, E.; De Muynck, S.; Asselah, T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J. Hepatol. 2011, 55, 1150–1153. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.Y.; Chen, S.S. Autophagy: A novel guardian of HCV against innate immune response. Autophagy 2011, 7, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. Hepatitis C Virus Induces the Localization of Lipid Rafts to Autophagosomes for Its RNA Replication. J. Virol. 2017, 91, JVI-00541. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ou, J.J. Regulation of Apolipoprotein E Trafficking by Hepatitis C Virus-induced Autophagy. J. Virol. 2018. [Google Scholar] [CrossRef]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Qu, A.; Han, X.; Wang, Y. HCV core protein represses the apoptosis and improves the autophagy of human hepatocytes. Int. J. Clin. Exp. Med. 2015, 8, 15787–15793. [Google Scholar]
- Quan, M.; Liu, S.; Li, G.; Wang, Q.; Zhang, J.; Zhang, M.; Li, M.; Gao, P.; Feng, S.; Cheng, J. A functional role for NS5ATP9 in the induction of HCV NS5A-mediated autophagy. J. Viral Hepat. 2014, 21, 405–415. [Google Scholar] [CrossRef]
- Matsui, C.; Deng, L.; Minami, N.; Abe, T.; Koike, K.; Shoji, I. Hepatitis C Virus NS5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1alpha via Chaperone-Mediated Autophagy. J. Virol. 2018, 92, JVI-00639. [Google Scholar] [CrossRef]
- Ren, H.; Elgner, F.; Jiang, B.; Himmelsbach, K.; Medvedev, R.; Ploen, D.; Hildt, E. The Autophagosomal SNARE Protein Syntaxin 17 Is an Essential Factor for the Hepatitis C Virus Life Cycle. J. Virol. 2016, 90, 5989–6000. [Google Scholar] [CrossRef] [Green Version]
- Ran, M.; Chen, H.; Liang, B.; Liao, W.; Jiang, J.; Huang, J.; Ning, C.; Zang, N.; Zhou, B.; Liao, Y.; et al. Alcohol-induced autophagy via upregulation of PIASy promotes HCV replication in human hepatoma cells. Cell Death Dis. 2018, 9, 898. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Li, J.R.; Peng, Z.G.; Zhang, J.P. Differential Effects of Autophagy-Related 10 Protein on HCV Replication and Autophagy Flux Are Mediated by Its Cysteine(44) and Cysteine(135). Front. Immunol. 2018, 9, 2176. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, Z.Y.; Zhang, J.P.; Jiang, J.D.; Ma, Y.Y.; Li, J.R.; Peng, Z.G.; Chen, J.H. Dual Roles of Two Isoforms of Autophagy-related Gene ATG10 in HCV-Subgenomic replicon Mediated Autophagy Flux and Innate Immunity. Sci. Rep. 2017, 7, 11250. [Google Scholar] [CrossRef]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Kurt, R.; Chandra, P.K.; Aboulnasr, F.; Panigrahi, R.; Ferraris, P.; Aydin, Y.; Reiss, K.; Wu, T.; Balart, L.A.; Dash, S. Chaperone-Mediated Autophagy Targets IFNAR1 for Lysosomal Degradation in Free Fatty Acid Treated HCV Cell Culture. PLoS ONE 2015, 10, e0125962. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732. [Google Scholar] [CrossRef]
- Shinohara, Y.; Imajo, K.; Yoneda, M.; Tomeno, W.; Ogawa, Y.; Kirikoshi, H.; Funakoshi, K.; Ikeda, M.; Kato, N.; Nakajima, A.; et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 2013, 432, 326–332. [Google Scholar] [CrossRef]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef]
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog. 2017, 13, e1006609. [Google Scholar] [CrossRef]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef]
- Mori, H.; Fukuhara, T.; Ono, C.; Tamura, T.; Sato, A.; Fauzyah, Y.; Wada, M.; Okamoto, T.; Noda, T.; Yoshimori, T.; et al. Induction of selective autophagy in cells replicating hepatitis C virus genome. J. Gen. Virol. 2018. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshimura, K.; Tamura, H.; Ueno, T.; Nishimura, T.; Inoue, T.; Sasaki, M.; Koike, M.; Arai, H.; Kominami, E.; et al. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem. Biophys. Res. Commun. 2010, 393, 274–279. [Google Scholar] [CrossRef]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M. Potential role of p62 in tumor development. Autophagy 2011, 7, 1088–1090. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Greenwood, M.P.; Murphy, D.; Isidoro, C.; Auewarakul, P.; Smith, D.R. Induced autophagy reduces virus output in dengue infected monocytic cells. Virology 2011, 418, 74–84. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Linking dengue virus entry and translation/replication through amphisomes. Autophagy 2009, 5, 434–435. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Hu, H.Y.; Kuo, S.H.; Lei, H.Y.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Dengue virus infection induces autophagy: An in vivo study. J. Biomed. Sci. 2013, 20, 65. [Google Scholar] [CrossRef]
- Mateo, R.; Nagamine, C.M.; Spagnolo, J.; Mendez, E.; Rahe, M.; Gale, M., Jr.; Yuan, J.; Kirkegaard, K. Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 2013, 87, 1312–1321. [Google Scholar] [CrossRef]
- Fang, Y.T.; Wan, S.W.; Lu, Y.T.; Yao, J.H.; Lin, C.F.; Hsu, L.J.; Brown, M.G.; Marshall, J.S.; Anderson, R.; Lin, Y.S. Autophagy facilitates antibody-enhanced dengue virus infection in human pre-basophil/mast cells. PLoS ONE 2014, 9, e110655. [Google Scholar] [CrossRef]
- Wu, Y.W.; Mettling, C.; Wu, S.R.; Yu, C.Y.; Perng, G.C.; Lin, Y.S.; Lin, Y.L. Autophagy-associated dengue vesicles promote viral transmission avoiding antibody neutralization. Sci. Rep. 2016, 6, 32243. [Google Scholar] [CrossRef]
- Chu, L.W.; Huang, Y.L.; Lee, J.H.; Huang, L.Y.; Chen, W.J.; Lin, Y.H.; Chen, J.Y.; Xiang, R.; Lee, C.H.; Ping, Y.H. Single-virus tracking approach to reveal the interaction of Dengue virus with autophagy during the early stage of infection. J. Biomed. Opt. 2014, 19, 011018. [Google Scholar] [CrossRef]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodriguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue Virus Inhibition of Autophagic Flux and Dependency of Viral Replication on Proteasomal Degradation of the Autophagy Receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J. Virol. 2017, 91, JVI-02020. [Google Scholar] [CrossRef]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef]
- Chen, H.R.; Chuang, Y.C.; Lin, Y.S.; Liu, H.S.; Liu, C.C.; Perng, G.C.; Yeh, T.M. Dengue Virus Nonstructural Protein 1 Induces Vascular Leakage through Macrophage Migration Inhibitory Factor and Autophagy. PLoS Negl. Trop. Dis. 2016, 10, e0004828. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef] [Green Version]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef]
- Thepparit, C.; Khakpoor, A.; Khongwichit, S.; Wikan, N.; Fongsaran, C.; Chingsuwanrote, P.; Panraksa, P.; Smith, D.R. Dengue 2 infection of HepG2 liver cells results in endoplasmic reticulum stress and induction of multiple pathways of cell death. BMC Res. Notes 2013, 6, 372. [Google Scholar] [CrossRef]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar]
- Tasaki, T.; Nukuzuma, S.; Takegami, T. Impaired Japanese encephalitis virus replication in p62/SQSTM1 deficient mouse embryonic fibroblasts. Microbiol. Immunol. 2016, 60, 708–711. [Google Scholar] [CrossRef]
- Wang, X.; Hou, L.; Du, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Capsid, membrane and NS3 are the major viral proteins involved in autophagy induced by Japanese encephalitis virus. Vet. Microbiol. 2015, 178, 217–229. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, K.B.; Chauhan, S.; Bhattacharyya, S.; Vrati, S.; Kalia, M. Diphenyleneiodonium enhances oxidative stress and inhibits Japanese encephalitis virus induced autophagy and ER stress pathways. Biochem. Biophys. Res. Commun. 2018, 502, 232–237. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, N.; Chen, S.; Zhao, P.; Ren, H.; Zhu, S.; Tang, H.; Zhu, Y.; Qi, Z. E3 Ubiquitin Ligase Nedd4 Promotes Japanese Encephalitis Virus Replication by Suppressing Autophagy in Human Neuroblastoma Cells. Sci. Rep. 2017, 7, 45375. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef] [Green Version]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef]
- Kobayashi, S.; Orba, Y.; Yamaguchi, H.; Takahashi, K.; Sasaki, M.; Hasebe, R.; Kimura, T.; Sawa, H. Autophagy inhibits viral genome replication and gene expression stages in West Nile virus infection. Virus Res. 2014, 191, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, A.B.; Martin-Acebes, M.A.; Saiz, J.C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2014, 5, 797. [Google Scholar]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10, 259. [Google Scholar] [CrossRef]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68. [Google Scholar] [CrossRef]
- Mizui, T.; Yamashina, S.; Tanida, I.; Takei, Y.; Ueno, T.; Sakamoto, N.; Ikejima, K.; Kitamura, T.; Enomoto, N.; Sakai, T.; et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 2010, 45, 195–203. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; Muniz, J.A.; Imbeloni, A.A.; da Fonseca, B.A. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol. 2015, 28, 161–169. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; da Fonseca, B.A. Chloroquine inhibits dengue virus type 2 replication in Vero cells but not in C6/36 cells. Sci. World J. 2013, 2013, 282734. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.R.; Tsai, T.T.; Chen, C.L.; Jhan, M.K.; Tsai, C.C.; Lee, Y.C.; Chen, C.H.; Lin, C.F. Blockade of dengue virus infection and viral cytotoxicity in neuronal cells in vitro and in vivo by targeting endocytic pathways. Sci. Rep. 2017, 7, 6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.F.; Lin, Y.S.; Huang, N.C.; Yu, C.Y.; Tsai, W.L.; Chen, J.J.; Kubota, T.; Matsuoka, M.; Chen, S.R.; Yang, C.S.; et al. Hydroxychloroquine-inhibited dengue virus is associated with host defense machinery. J. Interferon Cytokine Res. 2015, 35, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Xu, Q.Q.; Wu, D.G.; Ren, H.; Zhao, P.; Lao, W.G.; Wang, Y.; Tao, Q.Y.; Qian, X.J.; Wei, Y.H.; et al. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J. Virol. 2012, 86, 13407–13422. [Google Scholar] [CrossRef]
- Andoh, T.; Kawamata, H.; Umatake, M.; Terasawa, K.; Takegami, T.; Ochiai, H. Effect of bafilomycin A1 on the growth of Japanese encephalitis virus in Vero cells. J. Neurovirol. 1998, 4, 627–631. [Google Scholar] [CrossRef]
- Delvecchio, R.; Higa, L.M.; Pezzuto, P.; Valadao, A.L.; Garcez, P.P.; Monteiro, F.L.; Loiola, E.C.; Dias, A.A.; Silva, F.J.; Aliota, M.T.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Mesci, P.; Pinto, A.; Fernandes, I.; Sheets, N.; Shresta, S.; Farhy, C.; Huang, C.T.; Strongin, A.Y.; Muotri, A.R.; et al. Repurposing of the anti-malaria drug chloroquine for Zika Virus treatment and prophylaxis. Sci. Rep. 2017, 7, 15771. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Borges, M.C.; Castro, L.A.; Fonseca, B.A. Chloroquine use improves dengue-related symptoms. Memórias do Instituto Oswaldo Cruz 2013, 108, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Canete, R.; Rivas, D.E.; Escobedo, A.A.; Gonzalez, M.E.; Almirall, P.; Brito, K. A randomized, controlled, open-label trial evaluating the efficacy and safety of chloroquine in the treatment of giardiasis in children. West Indian Med. J. 2010, 59, 607–611. [Google Scholar]
- Tricou, V.; Minh, N.N.; Van, T.P.; Lee, S.J.; Farrar, J.; Wills, B.; Tran, H.T.; Simmons, C.P. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis. 2010, 4, e785. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Homeostatic Role of Autophagy in Hepatocytes. Semin. Liver Dis. 2018, 38, 308–319. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of autophagy. Three types of autophagy, including macroautophagy, microautophagy and chaperone-mediated autophagy, have been identified. Macroautophagy undergoes a stepwise vacuole biogenesis process that sequestrates the intracellular components within autophagosomes, which finally fuses with lysosomes to degrade the engulfed cargoes. Microautophagy is an invagination and scission process of lysosomal membrane that directly engulfs the intracellular portions to the lumen of lysosomes for degradation. Chaperone-mediated autophagy (CMA) involves the recognition of substrates that contain KFERQ motifs via the heat shock cognate protein of 70 KDa (Hsc70) and their delivery into the lysosomal lumen through the lysosomal membrane protein 2A (LAMP2A). For macroautophagy, two kinds of metabolic sensors, the mammalian target of rapamycin (mTOR) complex and 5′-AMP-activated protein kinase (AMPK) differentially regulate autophagy initiation. When cells are starved of nutrients, AMPK act as a positive regulator to activate autophagy through inhibiting mTOR. The inhibition of mTOR leads to translocation of unc-51 like-kinase (ULK) complex (ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101) to autophagy initiation site. Then, the ULK complex recruits and activates the class III phosphatidylinositol-3-OH kinase (class III-PI3K complex, including Vps34/PI3KC3, Vps15, Beclin 1 and ATG14) to generate PtdIn(3)P. The newly synthesized PtdIn(3)P recruits the double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI) family proteins to form the isolation membrane (IM)/phagophore. Two ubiquitin-like (UBL) conjugation systems are required for the expansion and elongation of phagophore to form autophagosomes. The ubiquitin conjugation enzyme 1 (E1) ATG7 activates ATG12 via the formation of a thioester bond between the C-terminal glycine of ATG12 and the cysteine residue of ATG7. Then ATG12 is transferred to ATG10 (E2) and subsequently conjugated to ATG5, yielding an ATG5-ATG12 complex. Finally, the ATG12-ATG5 conjugate interacts with ATG16L to form an ATG12-ATG5-ATG16L complex. ATG8/LC3 family proteins are cleaved by a cysteine protease ATG4 to generate the ATG8/LC3-I. Then ATG8/LC3-I is covalently linked to phosphatidylethanolamine (PE) to form the lipidated form of LC3 (ATG8/LC3-II) through enzymatic reactions of the ATG7 E1 and ATG3 E2. The mature autophagosomes fuse with lysosomes to form autolysosomes, in which the sequestrated materials are degraded. The small GTPase Ras-related protein 7 (Rab7) regulates the fusion of autophagosomes with lysosomesby interacting with cytoskeleton-associated factors, the FYVE and coiled-coil domain-containing 1 (FYCO1) and Rab-interacting lysosomal protein (RILP). Moreover, the concerted actions of multiple proteins on the HOPS complex, including sytaxin17 (STX17), the UV radiation resistance-associated (UVRAG), ATG14 and the pleckstrin homology domain-containing protein family member 1 (PLEKHM1) also participate in the maturation process of autolysosome.
Figure 1.
Overview of autophagy. Three types of autophagy, including macroautophagy, microautophagy and chaperone-mediated autophagy, have been identified. Macroautophagy undergoes a stepwise vacuole biogenesis process that sequestrates the intracellular components within autophagosomes, which finally fuses with lysosomes to degrade the engulfed cargoes. Microautophagy is an invagination and scission process of lysosomal membrane that directly engulfs the intracellular portions to the lumen of lysosomes for degradation. Chaperone-mediated autophagy (CMA) involves the recognition of substrates that contain KFERQ motifs via the heat shock cognate protein of 70 KDa (Hsc70) and their delivery into the lysosomal lumen through the lysosomal membrane protein 2A (LAMP2A). For macroautophagy, two kinds of metabolic sensors, the mammalian target of rapamycin (mTOR) complex and 5′-AMP-activated protein kinase (AMPK) differentially regulate autophagy initiation. When cells are starved of nutrients, AMPK act as a positive regulator to activate autophagy through inhibiting mTOR. The inhibition of mTOR leads to translocation of unc-51 like-kinase (ULK) complex (ULK1/2, ATG13, RB1-inducible coiled-coil 1 (RB1CC1, also known as FIP200) and ATG101) to autophagy initiation site. Then, the ULK complex recruits and activates the class III phosphatidylinositol-3-OH kinase (class III-PI3K complex, including Vps34/PI3KC3, Vps15, Beclin 1 and ATG14) to generate PtdIn(3)P. The newly synthesized PtdIn(3)P recruits the double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI) family proteins to form the isolation membrane (IM)/phagophore. Two ubiquitin-like (UBL) conjugation systems are required for the expansion and elongation of phagophore to form autophagosomes. The ubiquitin conjugation enzyme 1 (E1) ATG7 activates ATG12 via the formation of a thioester bond between the C-terminal glycine of ATG12 and the cysteine residue of ATG7. Then ATG12 is transferred to ATG10 (E2) and subsequently conjugated to ATG5, yielding an ATG5-ATG12 complex. Finally, the ATG12-ATG5 conjugate interacts with ATG16L to form an ATG12-ATG5-ATG16L complex. ATG8/LC3 family proteins are cleaved by a cysteine protease ATG4 to generate the ATG8/LC3-I. Then ATG8/LC3-I is covalently linked to phosphatidylethanolamine (PE) to form the lipidated form of LC3 (ATG8/LC3-II) through enzymatic reactions of the ATG7 E1 and ATG3 E2. The mature autophagosomes fuse with lysosomes to form autolysosomes, in which the sequestrated materials are degraded. The small GTPase Ras-related protein 7 (Rab7) regulates the fusion of autophagosomes with lysosomesby interacting with cytoskeleton-associated factors, the FYVE and coiled-coil domain-containing 1 (FYCO1) and Rab-interacting lysosomal protein (RILP). Moreover, the concerted actions of multiple proteins on the HOPS complex, including sytaxin17 (STX17), the UV radiation resistance-associated (UVRAG), ATG14 and the pleckstrin homology domain-containing protein family member 1 (PLEKHM1) also participate in the maturation process of autolysosome.

Figure 2.
Selective autophagy and cargo receptors. Selective autophagy involves the targeting of cargos to autophagic process via the receptor proteins that contains an ATG8/LC3-interacting regions (LIRs) for the binding to ATG8/LC3 located on the membrane of IM/phagophore. The autophagosomes elongated from IM/phagophore fuse with lysosomes to form autolysosomes, in which the engulfed cargos are degraded. The ubiquitination of cargo or an additional adaptor protein is often required for the recognition process between cargos and receptors proteins. Selective autophagy participates in the elimination of various kinds of organelles and proteins. Mammalian and yeast cargo receptors responsible for the degradation of the corresponding cargos by selective autophagy are listed as indicated. Selective autophagy participates in the degradations of damaged mitochondria, injured lysosomes, damaged peroxisomes, stressed endoplasmic reticulum (ER) and infected pathogens within autophagic degradation (mitophagy, lysophagy, pexophagy, ER-phagy and xenophagy, respectively). Moreover, other cargos, including lipid droplets (LDs) ferritin, nuclei, ribosomes, protein aggregates can be selectively sequestrated by selective autophagy for degradation via different types of cargo receptors.
Figure 2.
Selective autophagy and cargo receptors. Selective autophagy involves the targeting of cargos to autophagic process via the receptor proteins that contains an ATG8/LC3-interacting regions (LIRs) for the binding to ATG8/LC3 located on the membrane of IM/phagophore. The autophagosomes elongated from IM/phagophore fuse with lysosomes to form autolysosomes, in which the engulfed cargos are degraded. The ubiquitination of cargo or an additional adaptor protein is often required for the recognition process between cargos and receptors proteins. Selective autophagy participates in the elimination of various kinds of organelles and proteins. Mammalian and yeast cargo receptors responsible for the degradation of the corresponding cargos by selective autophagy are listed as indicated. Selective autophagy participates in the degradations of damaged mitochondria, injured lysosomes, damaged peroxisomes, stressed endoplasmic reticulum (ER) and infected pathogens within autophagic degradation (mitophagy, lysophagy, pexophagy, ER-phagy and xenophagy, respectively). Moreover, other cargos, including lipid droplets (LDs) ferritin, nuclei, ribosomes, protein aggregates can be selectively sequestrated by selective autophagy for degradation via different types of cargo receptors.


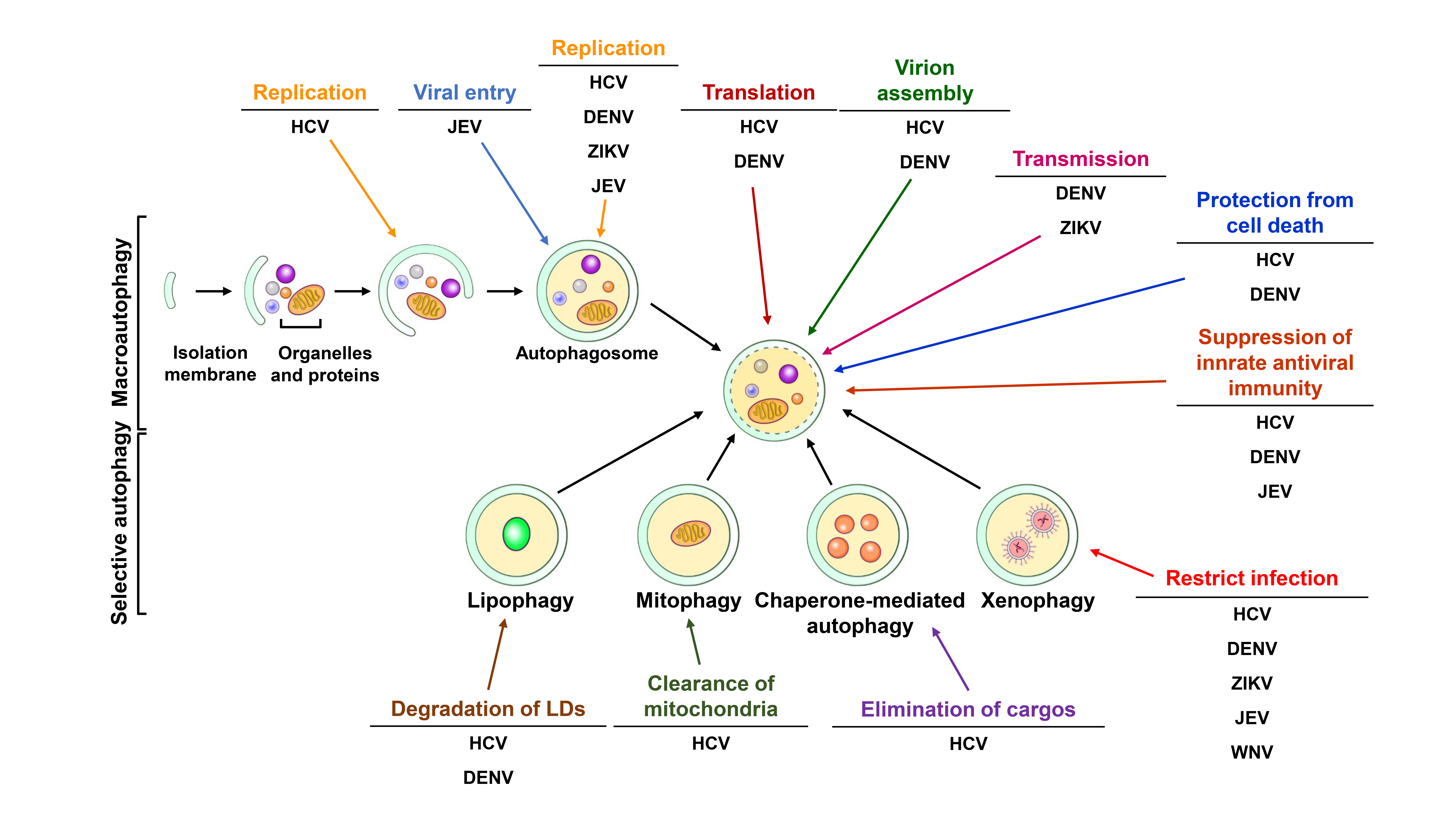{kind=link}
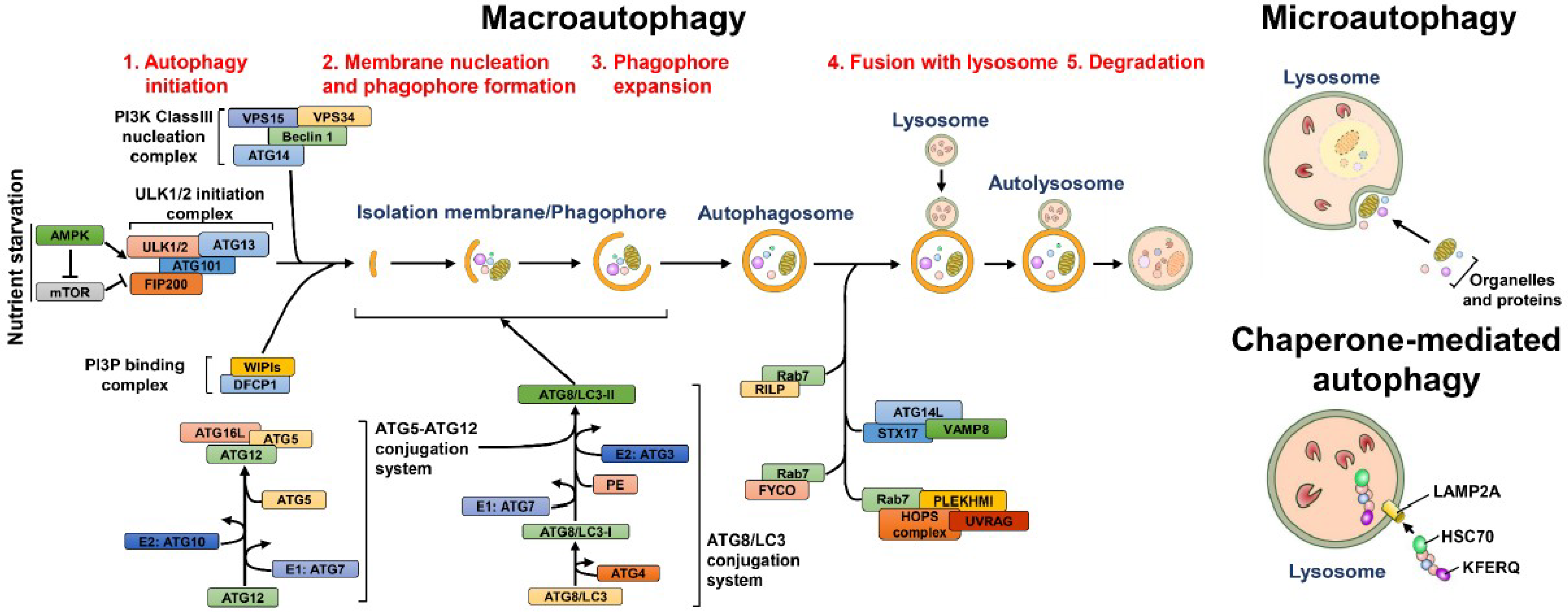{kind=link}
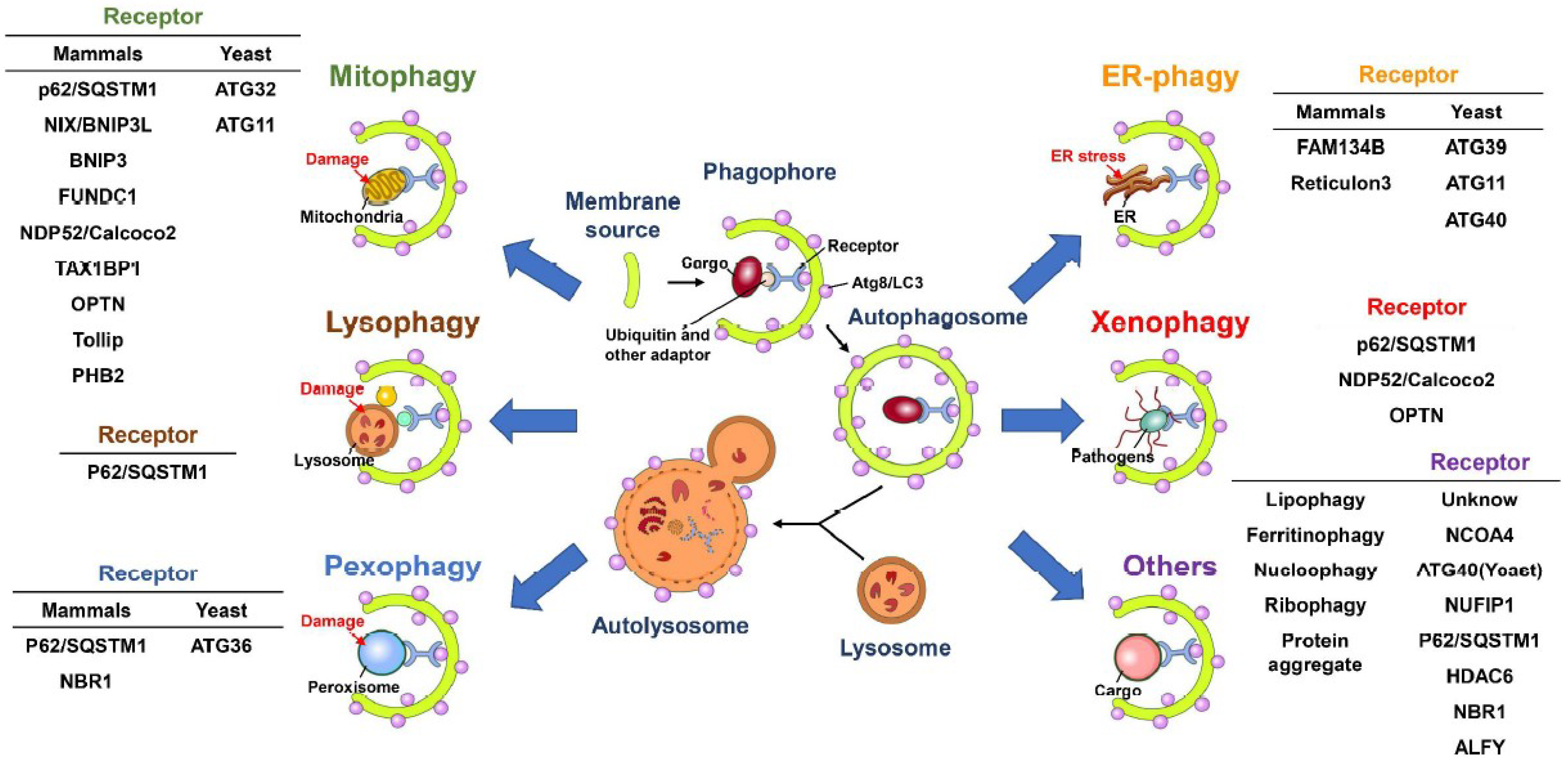{kind=link}
Table 1.
Summary of HCV-autophagy interactions.
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| H77 (1a) |
|
| Unknown | Ait-Goughoulte et al. [399] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Viral RNA transfection |
| Promotion on viral RNA replication | Sir et al. [401] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Dreux et al. [405] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Ke and Chen [406,410] |
| JFH1 (2a) |
|
|
| Shrivastava et al. [407] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Chan et al. [408] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Sir et al. [411] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Guevin et al. [412] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1cells/Transfection of replicon RNA |
|
| Ferraris et al. [413] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Kim et al. [414] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection |
| Promotion on the release of viral particles | Tanida et al. [415] |
|
|
| Promotion on the release of viral particles | Shrivastava et al. [416] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection and transfection of replicon RNA |
| Promotion on the release of viral particles | Kim et al. [417] |
| Human hepatocellular carcinoma, Huh7 cells/Transfection of replicon RNA |
| Counteracting the viral-induced cell death | Taguwa et al. [418] |
| JC1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Infection and transfection of viral proteins |
| Unknown | Su et al. [419] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Infection |
| Promotion on viral RNA replication | Gregoire et al. [420] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/ Transfection of viral proteins |
| Unknown | Wang et al. [421] Liu et al. [422] |
| Genotype 1b |
|
| Unknown | Quan et al. [423] |
| J6/JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection and transfection of viral proteins |
| Promotion on HNF1α degradation | Matsui et al. [424] |
| Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Transfection of viral RNA |
| Facilitation on virion release | Ren et al. [425] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Wang et al. [424] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
| Promotion on viral RNA replication by alcohol-induced autophagy | Ran et al. [426] |
| J4L6s (1b) | Human hepatocellular carcinoma, HepG2 cells/Transfection of replicon RNA |
| Promotion on the degradation of replicon RNA | Zhao et al. and Zhang et al. [427,428] |
| 1. Con1 (1b) 2. JFH1 (1a) |
|
| Promotion on LDs catabolism | Vescovo et al. [429] |
| 1. JFH1 (2a) 2. BM4–5 Feo (1b) | Human hepatocellular carcinoma, Huh7.5.1 cells /Infection and transfection of replicon RNA |
|
| Kim et al. [430,431] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells /Infection and transfection of viral protein |
| Sustained mitochondrial injury | Hara et al. [432] |
| 1. JFH1 (2a) 2. Con1 (1b) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection and transfection of replicon RNA |
| Unknown | Mori et al. [424] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection |
| Suppression of innate antiviral immunity | Kurt el al. [433] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Kim et al. [434] |
Table 2.
Summary of DENV-autophagy interactions.
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| DENV-2 (PL046) |
|
| Promotion on viral RNA replication | Lee et al. [398] |
| DENV-2 (16681) |
|
| Restriction on virus production | Panyasrivanit et al. [462] |
| DENV-2 (16681) |
|
|
| Heaton et al. [463] |
|
|
| Promotion on viral RNA replication | Khakpoor et al. [464] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
| Support on viral RNA replication and translation | Panyasrivanit et al. [465,466] |
| DENV-2 (PL046) | ICR mice |
| Promotion on virus replication and disease progression in vivo | Lee et al. [467] |
|
|
|
| Mateo et al. [468] |
| DENV-2 (16681) |
|
| Facilitation of DENV ADE infection | Fang et al. [469] |
| 1. Human monocytic cell line, U937 cells /Infection 2. Mouse embryonic fibroblast, wild type and ATG5 knockout cells/Infection |
| Promotion on cell to cell transmission | Wu et al. [470] |
| DENV-2 (16681) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection |
| Facilitation on virus entry | Chu et al. [471] |
| DENV-2 (NGC) | Human hepatocellular carcinoma, Huh7 cells/Infection |
| Promotion on virus replication | Metz et al. [472] |
| Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Zhang et al. [473] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Jordan and Randall [474] |
| DENV-2 (M544) | The Madin-Darby Canine Kidney, MDCK cells/Infection |
| Promotion on virus production | McLean et al. [475] |
| N/A | Human dermal microvascular endothelium, HMEC-1 cells/Infection |
| Promotion on disease pathogenesis | Chen et al. [476] |
| DENV-2 (PL046) |
|
|
| Lee et al. [477] |
| DENV-2 | The Madin-Darby Canine Kidney, MDCK cells/Infection |
|
| Datan et al. [478] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Thepparit et al. [479] |
Table 3.
Summary of JEV-autophagy interactions.
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
|
|
| Facilitation on the virion entry and uncoating | Li et al. [480] |
|
|
|
| Jin et al. [481] |
| JEV (JaGAr-01) strain | Mouse embryonic fibroblast, wild type and p62/SQSTM1 knockout cells/Infection |
| Promotion on viral RNA replication | Tasaki et al. [482] |
| JEV (SA14-14-2) strain |
|
| Promotion on viral RNA replication | Wang et al. [483] |
| JEV (P20778) strain |
|
| Inhibition on virus infection | Sharma et al. [484] |
| JEV (SA14-14-2) strain |
|
| Inhibition on virus infection | Xu et al. [486] |
| JEV (P20778) strain |
|
| Inhibition on virus production | Sharma et al. [487] |
Table 4.
Summary of WNV-autophagy interactions.
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
|
|
| Promotion on viral RNA replication (specifically by the early stage of autophagy) | Beatman et al. [488] |
|
|
| Unknown | Vandergaast et al. [489] |
|
|
| Inhibition the viral genome replication at late infection stage | Kobayashi et al. [490] |
| WNV TX02 train | The 5-day-old C57BL/6J mice |
| Inhibition of virus infection | Shoji-Kawata et al. [492] |
| WNV North American isolates | The kidney epithelial cells extracted from an African green monkey, Vero cells/Infection |
| Unknown | Blázquez et al. [493] |
Table 5.
Summary of ZIKV-autophagy interactions.
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| PF-25013-18 (French Polynesia, 2013) | Primary human dermal fibroblasts/Infection |
| Promotion on viral RNA replication | Hamel et al. [494] |
| Human fetal neural stem cells/Infection |
| Promotion on viral RNA replication | Liang et al. [495] |
| GZ01 (China, 2016) | Human umbilical vein endothelial cells/Infection |
| Promotion on viral RNA replication | Peng et al. [496] |
| The Brazilian strain (Paraiba, 2015) |
|
|
| Cao et al. [497] |
| MR766 (Uganda, 1947) | Drosophila melanogaster/Infection |
| Restriction of virus infection by activation of antiviral immunity | Liu et al. [498] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ke, P.-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123940
AMA Style
Ke P-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. International Journal of Molecular Sciences. 2018; 19(12):3940. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123940
Chicago/Turabian StyleKe, Po-Yuan. 2018. "The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions" International Journal of Molecular Sciences 19, no. 12: 3940. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123940
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.